VASKULITT (REV 034-052)
101 Polyarteritis nodosa (PAN), Juvenil PAN, ADA2 mangel (DADA2) (REV 034)
Ragnar Gunnarsson
Prosedyrekoder: 6-minutter gangtest: FYFX05. Intravenøs infusjon: WBGM00. Intravenøs infusjon med cytostatika: WBOC05. EKG: FPFE15
ATC koder (for legemiddelstatistikk): Prednisolon: H02A B06 Immunsuppressive legemidler: L04A A. Behandling med cyklofosfamid: L01AA01.
Definisjon
Systemisk PAN er en alvorlig, sjelden nekrotiserende arteritt, som affiserer små og mellomstore arterier. PAN rammer ikke kapillærer eller små vener og er ikke assosiert til anti-neutrofil cytoplasma antistoffer (ANCA). PAN gir ikke glomerulonefritt (GN), men kan gi vaskulær nefropati med hypertensjon og/eller fallende nyrefunksjon. PAN affiserer generelt ikke kar i lungevev. PAN deles inn i primær og sekundær form. Sekundær form av PAN er assosiert til hepatitt B virus (HBV) infeksjon.
Historikk
PAN ble første gang beskrevet professor Adolf Kussmaul (1822-1902) og Rudolf Maier (1824-1888) i Tyskland i 1866. Ordet «polyarteritt» er beskrivende og viser til arteritt i flere arterier. Ordet «nodosa» er gitt etter knuteformete forandringer (dvs. aneurismer) ved palpasjon av affiserte arterier ved obduksjon.
Patogenese og patologi
Sykdomsårsaken er ukjent. Det er en sterk assosiasjon ved PAN og hepatitt B virus (HBV) infeksjon. Det er derimot kun en liten andel (ca. 1%) av HBV smittete som får PAN, men risikoen er mangedoblet av å få PAN (ca. 1000x) når man er blir smittet av HBV. PAN oppstår da vanligvis tidlig, innen par måneder fra en er smittet. Det er fortsatt uklarhet hvordan HBV forårsaker PAN. Det er rapportert at det foregår virusreplikasjon i kar og endotel skade og det dannes immunkomplekser som faller ut i kar og komplement aktivering. Det har vært enkeltstående rapporter om PAN i sammenheng med infeksjon ved hepatitt C virus (HCV). Det er også påvist assosiasjon mellom PAN og hårcelleleukemi som er en undergruppe av kronisk lymfatisk leukemi som utgår fra B-lymfocytter.
Det finnes ingen pålitelig dyremodell for patogenese av PAN og dette har åpenbart hemmet vår forståelse av patogenesen.
Histologisk ses betennelsesreaksjon i karvegg med fibrinoid nekrose i media-delen av karet og invasjon av makrofager, neutrofile, lymfocytter, med særlig CD8+ T-lymfocytter og eosinofile. Dette medfører til ødeleggelse av lamina elastica-laget i karet og det utvikles aneurismeutvidelser og tromboser i karveggen. Det er alltid fravær av granulom-betennelse og kjempeceller ved PAN. Etter hvert kommer arrdannelse med økende nedsettelse av fibrin, som leder til snevring av karlumen og stenosedannelse.
Kriterier for klassifikasjon og mål på sykdomsaktivitet
American College of Rheumatology (ACR) publiserte nå for 30 år tilbake, i 1990, klassifikasjonskriteria for PAN. Det er vært å minne på at den skiller ikke imellom PAN og mikroskopisk polyangiit (MPA) (Tabell 1) (1). Det krever minst 3 av 10 kriteria og ut fra dette anført sensitivitet på 82% og spesifisitet på 87% for PAN.
Tabell 1: American College of Rheumatology (ACR) 1990 klassifikasjonskriterier for polyarteritis nodosa.
Krever minst 3 av 10 kriteriene og da antatt sensitivitet på 82% og spesifisitet på 87% for PAN.
- Vekttap > 4kg (fra sykdomsdebut som ikke kan forklares med redusert matinntak)
- Livedo retikularis utslett
- Testes smerter
- Myalgi eller muskelsvakhet
- Mononeuropati eller polynevropati
- Diastolisk blodtrykk > 90mmHg
- Økt BUN (blood urea nitrogen) (>14,3 µmol/L) eller kreatinin (>132 µmol/L)
- Hepatitt B positivitet (pos HBs antigen eller antistoffer i serum)
- Abnormal arteriografi (m/ mikroaneurismer eller okklusjon)
- Positiv biopsi av små eller mellomstor arterie med neutrofile i karveggen
Chapel Hill Nomenaclature som kartlagte vaskulittsykdommer var presentert i 1994. De siste og reviderte International Chapel Hill Consensus Conference Nomenclature of Vasculitides (CHCC) kriteria ble utformet på internasjonal kongress på universitet i Chapel Hill i delstaten North-Caroline 2012 og ble presentert i 2013 ofte referert til CHCC (2).
Illustrasjon: Cho KH – Annals of dermatology (2012). CC BY- NC 3.0.
. CC BY- NC 3.0")
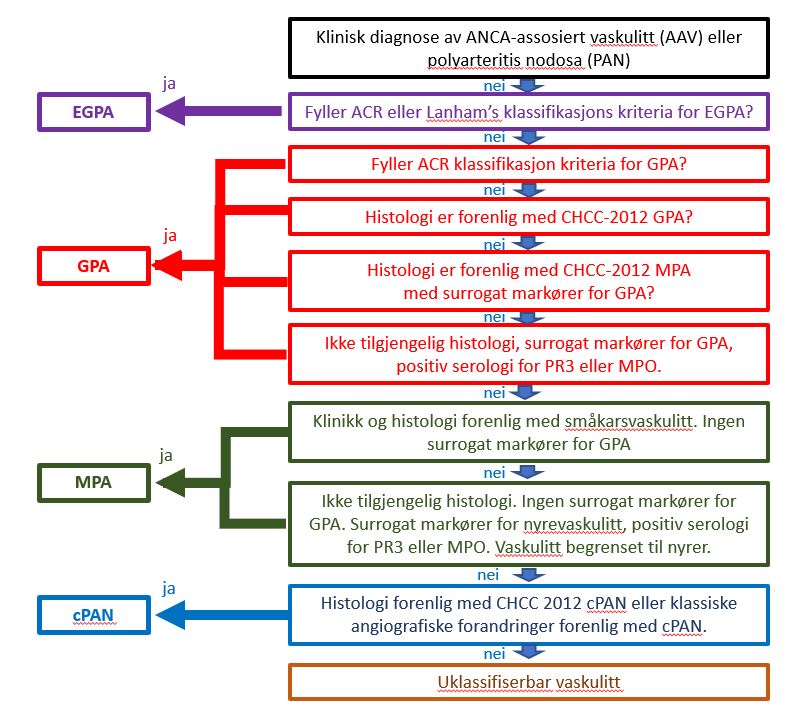
For å rasjonalisere klassifisering av forskjellige former av vaskulitter er anbefalt bl.a. av Det europeiske legemiddelbyrå (European Medicines Agency (EMA)) å anvende en algoritme med beslutnings-tre publisert i 2007, der PAN ligger nederst (Figur 1) (3).
Birmingham vasculitis index (BVAS) nå i tredje versjon, brukt som generell aktivitetsscore av vaskulitt og er anbefalt av European Vasculitis Study Group (EUVAS). Dette til tross det er best dokumentert ved ANCA assosierte vaskulitter (AAV) (4, 5). Det har også vært påvist at BVAS er et validert mål ved PAN hos barn og ungdommer (6).
Den franske såkalte “five-factor score” (FFS) kan utnyttes ofte i valg av behandling og har assosiasjon til prognose. Fleste bruker versjonen fra 1996 (FFS-Guïllevin-96) for PAN, som inkluderer de fem faktorene; (i) proteinuri > 1 g/døgn, (ii) nyresvikt (kreatinin > 140 µmol/L), (iii) kardiomyopati, (iv) gastrointestinal manifestasjoner, og (v) sentralnervøs affeksjon (7). Undertegnet ber folk være oppmerksomme på at det finnes to versjoner av FFS både (FFS-Guïllevin-96) og (FFS-Guïllevin fra 2011) som deler de samme tre av fem faktorene (ii-vi). Den senere versjonen av FFS (FFS-Guïllevin-11) er optimalisert for eosinofil granulomatøs polyangiit (EGPA) da har man tatt ut (i) proteinuri og byttet den ut med alder >65 år, økt grense for nyresvikt (ii) til kreatinin > 150 µmol/l og tatt ut sentralnervøse affeksjoner (v) og erstattet det med manglende symptomer øre, nese hals, som medfører at nærmest alle pasienter med PAN har FFS ≥ 1 ved FFS-Guïllevin 2011 versjonen (8).
Tabell 1. Five-factor score for eosinofil granulomatose med polyangiitt (EGPA). (7, 8)
| Revidert 2011 «five-factor score» (8) | 1996 «five-factor score» (7) |
| Alder > 65 år | Sentralnervesystem affeksjon |
| Manglende øre-nese hals affeksjon | Proteinuri (>1 g/dag) |
| Hjerteaffeksjon med hjertesvikt | Hjerteaffeksjon |
| Nyreaffeksjon med nyresvikt (serum kreatinin > 150 µmol/L (1,7mg/dL)) |
Nyreaffeksjon med nyresvikt (serum kreatinin > 140 µmmol/L (1,6mg/dL)) |
| Affeksjon av fordøyelsessystemet | Affeksjon av fordøyelsessystemet |
Figur 1. Foreslått EMA algoritme ved klassifikasjon av ANCA-assosiert vaskulitt og polyarteritis nodosa (3).
ACR: American College of Rheumatology; CHCC-2012 : Chapel Hill Consensus Conference 2012; cPAN : classic polyarteritis nodosa; EGPA : eosinofil granulomatose med polyangiitt; GPA : granulomatose med polyangiitt; MPA : mikroskopisk polyangiitt. MPO : myeloperoksidase; PR3 : proteinase 3.
Epidemiologi
PAN kan ramme pasienter i alle aldersgrupper og kan affisere barn, men opptrer hyppigst i aldersgruppen mellom 40-60 år og fleste kohorter viser at PAN rammer menn noe oftere.
PAN er nå en sjelden diagnose i Norge. I Tyskland har det vært rapportert svært lav årlig insidens på rundt 0,3 per million innbyggere (9, 10), mens undersøkelser fra Spania og England har vist betydelig høyere insidens på 6,2 og 9,7 per million innbyggere basert på ACR 1990 PAN kriteriene (11). Man har ingen gode epidemiologiske undersøkelser fra Norge på PAN, men inntrykket er at diagnosen er sjelden og sannsynlig nå nærmere de tyske tallene, en de engelske og spanske. Norge har også lav prevalens og for hepatitt B virusinfeksjon i befolkningen. Fallende insidens internasjonalt av PAN har noen forklaringer. Det første er at i eldre undersøkelser, spesielt før ANCA var tilgjengelig, og før Chapel Hill definisjonene (CHCC) ble utformet, var det et betydelig innslag av pasienter klassifisert med PAN, som per dags dato ville ha vært klassifisert med diagnosen mikroskopisk polyangiit (MPA). Det andre er at, i hvert fall i den vestlige verden, har antall hepatitt B falt dramatisk etter tilgjengelig behandling.
Nylig har kommet gentest til å fange opp vaskulitt assosiert til ADA2 mangel (DADA2) som uansett er sjelden diagnose og også genetiske vaskulære sykdommer som kan ha blitt tidligere vekslet med PAN det er ikke kun hos barn men kan også debutere i voksen alder og spesielt når dette assosieres til cerebrale manifestasjoner at man må tenke på ADA2 mangel der det har en særskilt behandling (12-14).
Juvenil PAN
Verken ACR eller CHCC er validert for barn (1, 2). Ved juvenil PAN er likt antall jenter og gutter rammet og gjennomsnittsalder er rundt 9 år (+/- 4 år) og insidens på under 1 per 100.000 per år. Sykdombildet er anført likt og hos voksne, men det er rapportert et høyere andel av hudaffeksjon ved systemisk form og av kutan PAN. Barn som gruppe ser ut til å ha bedre prognose en voksne, men får oftere tilbakefall (15). Tidligere undersøkelser har vist en sammenheng mellom streptokokk infeksjoner juvenil PAN.
Det er foreslått av SHARE (Single Hub and Access point for paediatric Rheumatology in Europe) å bruke Ankara 2008 kriteriene som ble utviklet for PAN hos barn og kalles EULAR/PReS klasifikasjonskriterier etter European League against Rheumatism / Peadiatric Rheumatology European Society for PAN (16-18). Disse inkluderer minst et av følgende; histologi forenlig med PAN eller typisk angiografi. I tillegg minst et av følgende fem manifestasjoner; hudaffeksjon, myalgi/muskelømhet, hypertensjon, perifer nevropati eller nyreaffeksjon. Generelt gis samme behandling hos voksne og barn (se her under).
Det finnes genetisk familiær form av vaskulitt, som kan klassifiseres til PAN og rammer pasienter vanligvis i ung alder og er assosiert til ADA2 mangel (DADA2) (14, 19, 20). Tilstanden var først rapportert i 2014. Pasienter med ADA2 mangel har tidligere vært klassifisert med PAN, men de har en særskilt prognose og behandling fra systemisk PAN. I tillegg er stimulator av interferon genet (STING) som er assosiert til vaskulopati av spedbarn (SAVI –STING-associated vasculopathy with onset in infancy) (15, 21, 22).
Kutan PAN
Det finnes isolert hudversjon av PAN – kutan PAN som karakteriseres klinisk av noduli, livedo retikularis og sårutvikling. Histologisk vises leukocytoklastisk vaskulitt med pannikulitt. Det foreligger sjelden andre organmanifestasjoner. Prognosen er generelt god og for kutan PAN og den regnes ikke som systemisk PAN og blir ikke referert her videre der behandlingen blir hovedsakelig i regi av hudleger.
Lokalisert PAN
Det er også sjelden form av monoorgan affeksjon av PAN som også kalles lokalisert PAN. Det har vært rapportert å affisere; galleblære, blindtarm og uterus og sjeldnere andre organer uten systemisk affeksjon. Ofte blir dette tilfeldig oppdaget ved operasjon og krever ikke immunsupresjons-behandling, men krever oppfølging der det er rapportert at opp til ¼ av disse utvikler systemisk PAN.
Klinikk
. CC BY-NC-ND 4.0")
Allmennsymptomer. Symptomdebut medfører som oftest generelle allmennsymptomer i form av feber og vekttap som rammer ca. 2/3. Myalgi (ca. 50%) og artralgi er relativ vanlig, men sjeldnere objektiv artritt. Sykdombilde og symptomer er selvfølgelig avhenge av hvilket kar er involverte.
Nerver. Affeksjon av perifer nervesystemet er også hyppig (60-74%) med utvikling av mononevritis multipleks, som lokaliseres ofte distalt og asymmetrisk og perifer nevropati som kommer oftest i løpet av de første månedene. Affeksjon av sentralnervesystemet sjelden (<5%) og kan affiserer cerebrale og spinale arterier med symptomer avhengig av lokalisasjon og om det foreligger aneurisme utvidelser, stenosering og/eller hematom. Kranial nevropati er sjelden (1%) og affiserer da som oftest nerver som forsyner øyemuskulatur dvs. III., IV. og VI. nerve. I tillegg til disse kan facialis nerven (VII.) og vestibulo-cochlear nerven (VIII.) bli rammet. Det er ikke vanlig med affeksjon av synsnerven (n. opticus) men det er vanligere med affeksjon at øyebunn med retinal vaskulitt, chorioretinitt, iritt og/eller iridocyklitt og man bør spørre og teste syn og synsfelt og ha en lav terskel for fundoskopisk undersøkelse og få øyelegetilsyn.
Akutt alvorlig tarmsykdom (tynntarm) er ingen sjelden manifestasjon ved PAN og affiserer ca. 40%-60%. Dette kan gi svært variert sykdomsbilde alt fra arteritt i blindtarm og/eller galleblære, eller kan gi akutt eller kronisk pankreatitt. Det kan gi infarkter og/eller blødninger i milt, sirkulasjonsforstyrrelser i tarm som kan gi ischemiske smerter ved matinntak. PAN kan også medføre gangren og perforasjon i tarm og/eller alvorlige gastrointestinal blødninger. Illustrasjon: Asti E, Pogliani L, Tritella S, Bonavina L – International journal of surgery case reports (2015). CC BY-NC-ND 4.0
Testes. PAN kan gi klaudikasjon smerter i legg og smerter i testikler med ischemisk orkitt.
Hudaffeksjon sees hos ca. 50% med livedo retikularis, livedo racemosa, ekkymoser eller palpabel purpura som oftest er 5-20 mm. store og kommer vanligvis distalt på underekstremiteter. Utvikling av distalt gangren og evt. sårutvikling og/eller splinter blødninger i neglesenger kan også forekomme.
Affeksjon av koronararterier kan forekomme og kan være lett å overse.
Nyreaffeksjon ved ischemisk nefropati kan gi nyresvikt og opptrer ofte tidlig i sykdomsfasen, men PAN gir ikke glomerulonefritt som mikroskopisk polyangiit. Isolert arteriell hypertensjon sees hos 35 % av pasienter med PAN. Mikroaneurisme i nyrearter og arterioler kan øke risiko for komplikasjoner ved nyrebiopsi med økt fare for blødningskomplikasjoner. (23).
Pasienter kan pga. vaskulær affeksjon av benvev utvikle beninfarkter og dette gir som oftest smertefulle leggsmerter og hevelse i legger og det kan sees periostal benaffeksjon på radiologiske undersøkelser.
Lungeaffeksjon ved PAN er som tidligere anført svært uvanlig og krever revurdering av PAN diagnosen evt. annen årsak for lungefunnene.
Undersøkelser
Generell klinisk undersøkelse samt rutene laboratorieundersøkelser inkl. inflammasjonsparametere, ANCA testing, screening for virushepatitt, serum elektroforese, kryoglobulin/kryofibrinogen og komplement-analyse og evt. også ANA screening.
Angiografi av abdominal kar kan vise stenosering og mikroaneurismer med det som kalles «rosenkrans tegn» (rosary sign).
Biopsi av nerve/muskel kan påvise affeksjon av epineural arterie og av muskelarterie. En dyp hudbiopsi kan påvise PAN.
Diagnosen
Klinisk mistanke om PAN oppstår oftest ved mønstergjenkjenning («pattern recognition») og man vil utrede den aktuelle pasient for aktuelle differensialdiagnoser (se herunder). Optimalt, er å stille diagnosen PAN på histologi, med funn av nekrotiserende arteritt med affeksjon av små og mellomstore arterier der det ofte sees ferske og eldre lesjoner i samme preparat. Noen pasienter kan ha histologiske tegn på PAN ved temporal biopsi. Dessverre er det ofte ikke mulig å bekrefte diagnosen histologisk. Mesenterial angiografi kan vise typiske radiologiske forandringer ved PAN mikroaneurismer og stenoser.
Differensialdiagnoser ved PAN
Mikroskopisk polyangiit (MPA) er hoveddifferensialdiagnosen og som anført her tidligere var MPA ofte klassifisert som PAN og flest alle studier og kohorter på PAN inkluderte oftest flertall de som vi i dag ville ha klassifisert som MPA. I tillegg er både granulomatøs polyangiit (GPA) og eosinofil granulomatøs polyangiit (EGPA) aktuelle differensialdiagnoser. Kryoglobulinemisk vaskulitt kan gi både nevropati og palpabel purpura og#chapter-eosinofil-granulomatos-polyangiitt-egpa-churg-strauss-vaskulitt nyreaffeksjon. Kawasaki sykdom (KD) en viktig differensialdiagnose, men både PAN og KD rammer mellomstore arterie der KD har en klar preferanse for koronarkar og rammer som oftest spedbarn eller unge barn.
Cancerassosiert vaskulitt er kan også være en aktuell differensialdiagnose ved PAN. Man har tidligere omhandlet tilstander som ADA2 mangel (DADA2) (gentest i Norge). Det er også andre monogenetiske vaskulære sykdommer som vaskulær Ehlers-Danlos (type IV), der det oftest (98%) foreligger mutasjon i genet COL3A1. Loeys-Dietz syndrom med fem forskjellige autosomal dominante genmutasjoner på gener som rammer Transforming Growth Factor beta (TGFBR1, TGFBR2, SMAD3, TGFB2 og TGFB3). Fibromuskulær dysplasi (FMD) og segmental arteriell mediolyse (SAM), kan være en viktige differensialdiagnoser men der følger som vanligvis ikke systemisk inflammasjon.
Vaskulopati/vaskulitt ved systemiske bindevevssykdommer som; systemisk lupus erythematosus (SLE), dermatomyositt (DM), mixed connective tissue disease (MCTD) og systemisk sklerose (SSc) samt og skleroderma/myositt overlapp syndromer samt og revmatoid vaskulitt.
Vaskulitt lignende tilstander som infeksjoner, inklusiv bakteriell endokarditt og vaskulopati ved antifosfolipid antistoffsyndrom med/uten Libman-Sachs endokarditt og andre non-bakterielle trombotiske endokarditter (NBTE) inklusiv myksom i hjerte.
Thrombangiitis obliterans (Buerger’s disease) og kolesterol embolier samt pyoderma gangrenosum (PG) kan være aktuelle differensialdiagnoser.
VEXAS syndrom bør vurderes i enkelte behandlingsresistente tilfeller hos menn over 45 år alder.
Behandling
Dessverre foreligger foreløpig svært få randomiserte behandlingsstudier på PAN. Eldre undersøkelser har stort innslag av pasienter med det som nå hadde vært klassifisert som MPA og i en del også EGPA og GPA og utgjør ofte MPA pasientene flertall av pasientene i mange av disse studiene. Behandlingen av PAN er lik behandling av ANCA assosierte vaskulitter (AAV), men det foreligger foreløpig lite dokumentasjon på rituksimab ved induksjonsbehandling ved PAN. Det er som anført mindre residiv fare ved PAN en AAV som medfører i fleste tilfeller kortere vedlikeholdsbehandling
Behandling med steroider (GC) godt dokumentert ved PAN. Før behandling med GC var fem-års overlevelse (5YSR) rundt 10%, men steg etter innføring av GC behandling til litt over 50%. Etter ytterligere innføring av cyklofosfamid behandling i tillegg steg 5YSR til over 80%.
Nevrogen affeksjon krever ofte behandling over lang tid. Tegn til bedring kan først vise seg 1-2 år etter debut av manifestasjonene og krever ofte smertestillendebehandling i tillegg.
Hepatitt B assosiert (HBV) PAN krever spesiell oppmerksomhet. I tillegg til induksjonsbehandling med glukokortikoider og cyklofosfamid er antiviral behandling viktig del av behandlingen, der det er stor risiko for økt virusreplikasjon ved immunsuppresjon, som øker risikoen for kronisk hepatitt og lever cirrhose. Målet er der å utvikle serumkonversjon for HBV og samtidig få kontroll over vaskulitt-sykdommen med å bruke minimal av immunsuppresjon.
Behandlingen av alvorlig PAN av alvorlig PAN og/eller økt FFS (FFS-Guïllevin-96 > 1) er vanligvis med induksjonsbehandling med høydose steroidbehandling sammen med cytotoksisk behandling enten p.o. cyklofosfamid eller i.v. puls cyklofosfamid behandling. Nylig utgitte behandlingsanbefalinger for PAN fra American College of Rheumatology (ACR) og Vasculitis Foundation og er alle de anbefalingene betingete («conditional») men ikke generelle pga. manglende dokumentasjon (24).
Fleste vil innlede behandling med å gi intravenøst metylprednisolon pulsbehandling 7,5 – 15 mg/kg (oftest 500 – 1.000 mg) der man gir dette oftest som daglige pulser over 3 dager etterfulgt av per oral glukokortikoid (GC) behandling. Oftest anvendes per oral Prednisolon behandling 1 mg/kg, men da oftest ikke mer enn 60 mg/d daglig og nedtrapping av behandlingen når det har kommet normalisering av inflammasjonsparametere (C-reaktiv protein). Prednisolon nedtrappingen og total behandlingslengde er dessverre ikke godt forskningsmessig dokumentert ved PAN.
Cyclofosfamid (CYC) som tilleggsbehandling til GC for alvorlig PAN er rimelig godt dokumentert (Grade 1B dokumentasjon). Pga. langtidsvirkninger, der høy kumulativ dosering av CYC er assosiert til økt kreft- og infeksjonsrisiko samt økt fare for infertilitet, ønsker man at anvende CYC i så kort tid og i så lav dosering som mulig. Intravenøs støtbehandling gir betydelig lavere kumulativ dosering en per oral behandling. Det er da viktig med god hydrering og forhindre at medikamentet blir stående i urinblæren. I tillegg anvender mange bruk av intravenøs og/eller per oral mesna/Uromitexan® som forebygging av urinveistoksisitet. Mesna binder seg til den toksiske oxazafosforinmetabolitten akrolein som reduserer dannelsen av akrolein i urinen og reduserer risikoen for hemoragisk cystitt. IV CYC behandlingen krever justering i henhold til alder og redusert nyrefunksjon (Tabel 2) som ved CYCLOPS/EUVAS cyklofosfamid protokollen ved AAV. Francais d’Etude des Vascularities (French Vasculitis Study Group) oftest kortet ned til FVSG har i egen protokoll for PAN som er består i CYC pulsbehandling 600 mg/m2 tilsvarer omtrent 15 mg/kg der fleste gir maksimalt intravenøst 1200 mg CYC om gangen. FVSG bruker å gi første tre doseringene med 2. ukers intervall, men de neste tre doseringene med fire ukers intervall (25). Andre har gitt IV CYC etter den såkalte CYCLOPS/EUVAS protokollen (26) som anvendes ofte ved AAV der man gir de første tre kurene, som ved FVSG protokollen, med to ukers intervall (i uke 0, 2, 4) og deretter neste tre med tre ukers intervall (uke 7, 10 og 13). Om dette gjør noen real forskjell kan man klart tvile på. Hvis pasienten fortsatt ikke har kommet i remisjon kan man gi videre CYC kurer til pasienten kommer i remisjon. Alternativt kan man vurdere å skifte fra intravenøs til per oral CYC behandling. 2021 ACR/VF retningslinjene går ikke inn på dosering eller behandlingslengde av cyklofosfamid (24).
Tabell 2 Doseendring av cyklofosfamid relatert til alder / nyrefunksjon
| CYC i.v. puls (mg/kg)(Max 1200mg) | ||
| Alder (år) | Kreatinin. ≤ 300(μmol/L) eller eGFR > 30 (ml/min/1,73m2) | Kreat. > 300eGFR: ≤ 30 |
| < 60 | 15 | 12,5 |
| 60 – 70 | 12,5 | 10,0 |
| > 70 | 10,0 | 7,5 |
Man har ved behandling av PAN overført en del av behandlingsprinsippene som er dokumentert ved AAV men det er vert å nevne at dokumentasjonen på steroidsparende effekten av annen immunsuppresjonsbehandling er per dags dato betydelig svakere enn ved AAV. Tradisjonelt brukes per oral daglig azathioprin (AZA) eller ukentlig per oral eller subkutant metotreksat (MTX) eller eventuelt som mindre dokumentert alternativt per oral mykofenolat mofetil (MMF) behandling. Vanligvis gis AZA eller MTX, alternativt MMF i rundt 18 måneder sammen med lavdose steroid behandling som vedlikehold etter induksjonsbehandling med CYC etter pasienten har kommet i remisjon.
ACR/VF anbefaler i sine retningslinjer å kombinere med enten MTX eller AZA med GC ved mindre alvorlig PAN foran å gi GC som monoterapi (24). Om GC behandlingen skal fullstendig fases ut eller om man skal bruke lavdose over tid er mer eller mindre overlatt til behandlende lege der dokumentasjon ikke finnes. Behandling utover 18 måneder er ikke særlig godt dokumentert, men det finnes pasienter man må vurdere økt behandlingslengde.
Azathioprin (AZA) – Imurel® som oftest rundt 2 mg/kg i en enkel dosering. Imurel finnes i 50 mg og 25 tabletter og det går ikke å dele dem. Man anbefaler å ta tiopurin methyltransferase (TPMT) genotype forut behandling. De fleste (rundt 90 %) har TPMT*1/*1 (wild-type) genotype med normal TPMT enzym aktivitet og kan få vanlig dosering, 2,0-2,5 mg/kg per dag en gang på dagen. Pasienter som er heterozygote med TPMT*1 og noen av de over 20 TPMT polymorfisme, som oftest er *2, *3A, *3B, *3C, og *4, har redusert TPMT enzym aktivitet og må få redusert, oftest halvert dosering og følges grundig hvis oppstart. De som ikke har TPMT*1genotypen og enten homozygot og eller heterozygot for en av de allelene bør ikke få azathioprin i hele tatt pga. økt risiko for alvorlig myelosuppresjon. Dette utgjør få pasienter (>1%). Vises til Avdeling for farmakologi. Oslo universitetssykehus. (https://anx.no/tpmt/)
Verdi av 6-tioguaninnukleotide (6-TGN) og metyl-merkaptopurin (me-MP) måles i heparinisert fullblod, tilsier om effekt av AZA behandlingen. Terapeutisk område er: 6-TGN 3,5−5,0 µmol/L og me-MP <50 µmol/L når brukes i transplantasjonsmedisin. Dette kan anvendes i kontroll med blodprøve. (https://anx.no/6tgn/)
Metotreksat (MTX) p.o eller s.c. i ukentlig dosering er medikament som brukes i et stort omfang innen revmatologien noe alle revmatologer er og bør være kjent med. Generelt er MTX s.c. er å foretrekke, der det gir mer stabil blodverdi. Vanlig dosering er opp mot ca. 0,3 mg/kg/uke (15-25 mg) og ofte trapper man gradvis opp mot måldosering over noen uker. Det har vært tradisjon i Europa å gi samtidig folsyre tilskudd for å redusere bivirkninger ofte 1 mg Folsyre daglig.
Mykofenolat mofetil (MMF) kan vurderes ved PAN. Dokumentasjon ved bruk av MMF ved PAN er svak. Dosering: 2-3 g/d fordelt på to doseringer, med ca. 12 timers intervall. Det er tabletter på 500 mg og kapsler på 250mg i tillegg finnes MMF i mikstur.
Det finnes enkeltkasuistikker om bruk av rituksimab (RTX) i kombinasjon med GC som behandling for PAN, men fleste inklusivt ACR og VF i sine retningslinjer anbefaler primært cyklofosfamid foran RTX (24).
Hos pasienter med ADA2 mangel (DADA2) anbefales sterkt å bruke tumor nekrose faktor alfa inhibitor (TNFi) som ser ut til å hindre cerebral affeksjon og utvikling til å få slag eller hjerneblødning (14, 19, 20, 24).
Prognose
Ubehandlet har PAN som oftest dårlig prognose og eldre retrospektive undersøkelser viser overlevelse på rundt 12-13%. Fem års dødeligheten ved PAN ved FFS (Guïllevin-1996) = 0 er 12 %, ved FFS = 1 er 26 % og ved FFS = 2 er 5 års dødelighet på 46 % (7). Som anført ut fra franske retningslinjer anvendes FFS ved valg av behandling men FFS gir i grunn prospektivt informasjon.
Av dødsårsaker av PAN dominerer myokardinfarkt, gastrointestinal blødninger, infarkter og perforasjon samt og cerebrovaskulære hendelser.
Fem-års-overlevelse (5YSR) i en stor fransk oppfølgingsundersøkelse fra French Vasculitis Study Group, med 348 pasienter inkluderte, viste at pasienter diagnostisert etter 1995, hadde ett-års-overlevelse (1YSR) på 97% og fem-års-overlevelse (5YSR) på 88%. Negative prognostiske faktorer var; høy alder, vekttap ≥ 5kg, utvikling av hypertensjon og gastrointestinal affeksjon av PAN samt «five-factor score» FSS ≥ 1 (23).
Det er relative sjelden at PAN pasienter får tilbakefall i hvert fall hvis man sammenligner med AAV pasienter og PAN er som oftest er monofasisk «one shot disease». I den retrospektive franske oppfølgingsstudien fikk 28% av HBV negative PAN pasienter versus 11% av HBV positive PAN pasienter tilbakefall (23). De HBV positive PAN som fikk tilbakefall hadde som oftest en vedvarende hepatitt B virusreplikasjon. Ved tilbakefall trenger sykdombildet ikke å være helt likt, men oftest noe mildere.
Det foreligger lite data på langvarig vedlikeholdsbehandling, som er neppe indisert i det store flertall av pasienter. Det foreligger ingen dokumentasjon på økt cancerrisiko hos de som har PAN uten HBV.
Referanser
1. Lightfoot RW, Jr., Michel BA, Bloch DA, Hunder GG, Zvaifler NJ, McShane DJ, et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum. 1990 Aug;33(8):1088-93. PubMed PMID: 1975174.
2. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013 Jan;65(1):1-11. PubMed PMID: 23045170.
3. Watts R, Lane S, Hanslik T, Hauser T, Hellmich B, Koldingsnes W, et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis. 2007 Feb;66(2):222-7. PubMed PMID: 16901958. PMCID: PMC1798520.
4. Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, Emery P, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM. 1994 Nov;87(11):671-8. PubMed PMID: 7820541.
5. Seror R, Bowman SJ, Brito-Zeron P, Theander E, Bootsma H, Tzioufas A, et al. EULAR Sjogren’s syndrome disease activity index (ESSDAI): a user guide. RMD Open. 2015 February 1, 2015;1(1):e000022. PubMed PMID: 26509054. PMCID: PMC4613159.
6. Demirkaya E, Ozen S, Pistorio A, Galasso R, Ravelli A, Hasija R, et al. Performance of Birmingham Vasculitis Activity Score and disease extent index in childhood vasculitides. Clin Exp Rheumatol. 2012 Jan-Feb;30(1 Suppl 70):S162-8. PubMed PMID: 22640658.
7. Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore). 1996 Jan;75(1):17-28. PubMed PMID: 8569467.
8. Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P. The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore). 2011 Jan;90(1):19-27. PubMed PMID: 21200183.
9. Reinhold-Keller E, Herlyn K, Wagner-Bastmeyer R, Gross WL. Stable incidence of primary systemic vasculitides over five years: results from the German vasculitis register. Arthritis Rheum. 2005 Feb 15;53(1):93-9. PubMed PMID: 15696553.
10. Reinhold-Keller E, Herlyn K, Wagner-Bastmeyer R, Gutfleisch J, Peter HH, Raspe HH, et al. No difference in the incidences of vasculitides between north and south Germany: first results of the German vasculitis register. Rheumatology (Oxford). 2002 May;41(5):540-9. PubMed PMID: 12011378.
11. Watts RA, Lane SE, Scott DG, Koldingsnes W, Nossent H, Gonzalez-Gay MA, et al. Epidemiology of vasculitis in Europe. Ann Rheum Dis. 2001 Dec;60(12):1156-7. PubMed PMID: 11760724. PMCID: PMC1753455.
12. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. The New England journal of medicine. 2014 Mar 6;370(10):921-31. PubMed PMID: 24552285.
13. Cooray S, Omyinmi E, Hong Y, Papadopoulou C, Harper L, Al-Abadi E, et al. Anti-tumour necrosis factor treatment for the prevention of ischaemic events in patients with deficiency of adenosine deaminase 2 (DADA2). Rheumatology (Oxford). 2021 Sep 1;60(9):4373-8. PubMed PMID: 33420503.
14. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. The New England journal of medicine. 2014 Mar 06;370(10):911-20. PubMed PMID: 24552284. PMCID: PMC4193683.
15. Ozen S, Sag E. Childhood vasculitis. Rheumatology (Oxford). 2020 May 1;59(Supplement_3):iii95-iii100. PubMed PMID: 32348513.
16. Ozen S, Ruperto N, Dillon MJ, Bagga A, Barron K, Davin JC, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006 Jul;65(7):936-41. PubMed PMID: 16322081. PMCID: PMC1798210.
17. Ruperto N, Ozen S, Pistorio A, Dolezalova P, Brogan P, Cabral DA, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part I: Overall methodology and clinical characterisation. Ann Rheum Dis. 2010 May;69(5):790-7. PubMed PMID: 20388738.
18. de Graeff N, Groot N, Brogan P, Ozen S, Avcin T, Bader-Meunier B, et al. European consensus-based recommendations for the diagnosis and treatment of rare paediatric vasculitides – the SHARE initiative. Rheumatology (Oxford). 2019 Apr 1;58(4):656-71. PubMed PMID: 30535249.
19. Aksentijevich I, Sampaio Moura N, Barron K. Adenosine Deaminase 2 Deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle
Copyright © 1993-2020, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 2020.
20. Huang Z, Li T, Nigrovic PA, Lee PY. Polyarteritis nodosa and deficiency of adenosine deaminase 2 – Shared genealogy, generations apart. Clin Immunol. 2020 Jun;215:108411. PubMed PMID: 32276138. PMCID: PMC7387119.
21. Tang X, Xu H, Zhou C, Peng Y, Liu H, Liu J, et al. STING-Associated Vasculopathy with Onset in Infancy in Three Children with New Clinical Aspect and Unsatisfactory Therapeutic Responses to Tofacitinib. J Clin Immunol. 2020 Jan;40(1):114-22. PubMed PMID: 31705453.
22. Jain A, Misra DP, Sharma A, Wakhlu A, Agarwal V, Negi VS. Vasculitis and vasculitis-like manifestations in monogenic autoinflammatory syndromes. Rheumatol Int. 2018 Jan;38(1):13-24. PubMed PMID: 29032440.
23. Pagnoux C, Seror R, Henegar C, Mahr A, Cohen P, Le Guern V, et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa: a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group Database. Arthritis Rheum. 2010 Feb;62(2):616-26. PubMed PMID: 20112401.
24. Chung SA, Gorelik M, Langford CA, Maz M, Abril A, Guyatt G, et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Polyarteritis Nodosa. Arthritis Rheumatol. 2021 Aug;73(8):1384-93. PubMed PMID: 34235883.
25. Samson M, Puéchal X, Mouthon L, Devilliers H, Cohen P, Bienvenu B, et al. Microscopic polyangiitis and non-HBV polyarteritis nodosa with poor-prognosis factors: 10-year results of the prospective CHUSPAN trial. Clin Exp Rheumatol. 2017 Mar-Apr;35 Suppl 103(1):176-84. PubMed PMID: 28422001.
26. de Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009 May 19;150(10):670-80. PubMed PMID: 19451574.
