BINDEVEVSSYKDOMMER (REV 021-033)
59
Myositt (Dermatomyositt og Polymyositt)
Jan Tore Gran and Øyvind Palm
Kjennetegn på inflammatorisk polymyositt og dermatomyositt
Progredierende svakhet i proksimale muskler (lår, overarmer).
Dermatomyositt (DM) med typiske hud-forandringer.
Vedvarende økt CK i blodet.
Noen med utslag i myosittspesifikke antistoff (myositt-blot).
MR av lårmuskler viser bilateralt muskelødem.
Non-inflammatorisk myopati og inklusjonslegeme myositt utelukkes.
Diagnosen sikres ved biopsi fra affisert muskulatur.
Diagnosekoder ICD-10: M33.1 (Dermatomyositt), M33.2 (Polymyositt). J99.1 Lungemanifestasjon ved bindevevssykdom; G72.0 Legemiddelindusert myositt/myopati; G72.8 Kreft-assosiert (annen) myopati
Prosedyrekoder: 6-minutter gangtest: FYFX05. Intravenøs infusjon: WBGM00. Behandling med rituksimab: L01XC02. Infusjon med gammaglobulin: RPGM05. EKG: FPFE15
ATC koder (for legemiddelstatistikk): L04A A Immunsuppressive legemidler
Definisjon
Myositt (idiopatisk inflammatorisk myopati) er en gruppe inflammatoriske revmatiske sykdommer. De kjennetegnes av inflammasjon og nekrose i tverrstripet muskulatur, og har ukjent sykdomsårsak. Typiske symptomer er progredierende svakhet i lår, overarmer og nakke, ofte uten særlig smerte. Også hud (dermatomyositt), lunge, hjerte og ledd kan være angrepet (Dourado E, 2023). Sykdommene deles vanligvis inn i fem grupper utenom overlapp-syndromer (Glaubitz S, 2020).
- Polymyositt (PM)
- Dermatomyositt (DM) (muskel- og hud-affeksjon) og juvenil dermatomyositt (JDM).
- Immunmediert nekrotiserende myopati (IMNM), inklusiv statin-indusert myopati deles inn i tre grupper: a) anti-HMGCA positiv type (statin-relatert hos ca. 50%), b) anti- SRP positiv type, c) antistoff negativ type.
- Antisyntetase syndrom
- Inklusjonslegememyositt (IBM)
I tillegg kommer kreft-assosiert myositt (CAM) (ledsages som oftest av hudaffeksjon), overlapp-syndromer (skleromyositt) og noen sjeldne undergrupper (andre, se sist i kapittelet).
Forskjell mellom myositt/inflammatorisk myopati skilles fra non-inflammatoriske myopatier er ulik anamnese, sykdomsutvikling, myositt har inflammatoriske forandringer med leukocyttinfiltrater i vevsprøve og ingen infeksjonstegn, mens non-inflammatorisk myopati preges av ikke-inflammatorisk nekrose, nevrogen atrofi eller sekundær inflammasjon (Barohn RJ, 2014).
Historie
- 1863 og 1887 Polymyositt og dermatomyositt beskrives av Ernst Leberecht Wagener (Keitel W , 2015)
- 1916 Dermatomyositt assosieres med malignitet (Tiniakou E, 2015)
- 1975 Klassifikasjon av subtyper av Peter & Bohan (Leclair V, 2018)
- 1990 Syv myosittspesifikke antistoff beskrives (Mi2, SRP, Jo-1, PL-7, PL-12, EJ, OJ)
- 2017 EULAR/ACR kriterier (Leclair V, 2018)
Sykdomsårsaker
Årsaken til myositt er stort sett ukjent, utenom at IMNM i noen tilfeller kan trigges av statiner. En viss genetisk disposisjon foreligger, men den er ikke sterk nok til å føre til en klar familiær disposisjon (Dourado E, 2023).
Epidemiologi
Prevalens 8-18/100 000, insidens 0,5-8 /mill./år. Hyppigst affiseres personer i 40-50 års alderen. Kvinner / Menn 2:1 (Dobloug G, 2013).
Symptomer og kliniske kjennetegn
Myositt kan ha en rekke ulike symptomer i starten, og det kan være vanskelig å knytte disse til diagnosen med en gang. Dette fører til at det i gjennomsnitt tar 2,25 år før riktig diagnosen stilles (Namsrai T, 2022). De første sykdomstegn kan være:
- Belastningsdyspne (antisyntetase syndrom) eller
- Eksantem (dermatomyositt)
- Forhøyet kreatin kinase (CK) av uklar årsak (spesielt ved inklusjonslegememyositt).
For en tidlig diagnose er det viktig at klinikeren i anamnesen er oppmerksom på de aktuelle symptomene og supplerer med aktuell utredning.

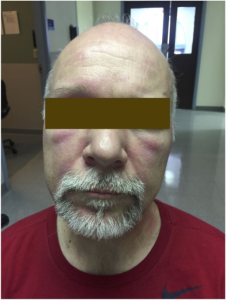
Dermatomyositt. 1) V-tegn på thoraks 2) Mekanikerhender 3) Gottrons papler over MCP, PIP og DIP-ledd 4) Sjal-tegn over nakke og skuldre. Illustrasjon: Mervi U, Indian J Derm, 2012. CC-BY-NC-SA 3.0
Allmenn-symptomer
Sykdomsfølelse, lett feber og vekttap forekommer. Mange har artralgier, særlig tidlig i sykdomsforløpet.
Muskler
Langsom progresjon. Muskelsymptomene utvikler seg gradvis, aldri i løpet av dager.
Symmetrisk kraftsvekkelse i proksimale muskler er typisk, oftest i nakke, skuldre hofter og lårmuskler. Unntaket er inklusjonslegeme myositt (IBM) som har mer distal affeksjon og ofte rammer underarmer og legger. Pasienten kan ha problemer med å reise seg fra en stol eller gå opp trapper uten støtte. Slitenhet, rask fysisk utmattelse og belastningsdyspne kan også være symptomer.
Svakhet i hoftemuskler påvirker gangfunksjonen og debuterer nesten alltid før affeksjonen av skulderbuen.
Smerten og stivhet er vanligvis beskjedne.
Nakke-fleksorer kan angripes, slik at uttalt svakhet medfører “drop-head” (differensialdiagnoser: amyloidose, nevrologiske sykdommer)
Svelg. Sjelden kan muskler i svelg rammes, noe som kan gi nasal tale, heshet og nese-regurgitasjon. Dysfagi skyldes affeksjon av tverrstripet muskulatur i farynks og øvre øsofagus.
Ubehandlet kan myositt medføre muskelatrofi, men uten myotoni eller myasteni.
Dermatomyositt. Når myositt er ledsaget av hudforandringer, klassifiseres tilstanden som dermatomyositt (DM). Hudsymptomene kan være kløende og lysømfintlig og følger ofte myositt-aktiviteten, men kan også debutere måneder etter muskel-affeksjonen.
Klinisk og histologisk kan dermatomyositt-utslett i ansikt eller hodebunn minne om lupus i huden (kutan lupus), men øvrige kliniske tegn og ved hjelp av antistoff bør disse tilstandene kunne skilles (Didona D, 2023).
-Gottrons tegn er erythem som sees på hendenes dorsale side, særlig over MCP og PIP leddene som rød-lilla forandringer, men kan også sees over albuer, knær.
-Gottrons papler er papulært erythematøst utslett, oftest over MCP-ledd, men de kan strekke seg over dorsalsiden av hele fingre (Ricceri F, 2015).
-V-tegn er et erythematøst, solsensitivt utslett på ansikt, hals og fremre bryst.
-Sjal (Shawl) -tegn betegner utslett på skuldre og øvre del av ryggen.
-Hylster/holster-tegn er et rødt-lilla, retikulært lateralt på kne og lår, der en slire ofte bæres (Cooper S, 2015).
-Poikiloderma består av teleangiektasier, hudatrofi og hypo- og hyper- pigmentering og inngår i Sjal-tegn, V-tegn og Hylster-tegn.
Mekaniker-hender (maskinist-hånd) er flassende, sprekk-dannelser på fingertupper, spesielt på radialsiden av pekefingre og på tommel. Dette sees oftest ved antisyntetase syndrom (vennligst se eget kapittel), men forekommer enkelte ganger hos pasienter med dermatomyositt.
Sjeldne hud-symptomer
- Subkutant ødem (uten proteinuri, lavt albumin eller andre tegn på nefritt) er assosiert med alvorlig sykdom. Ødem er vanligst ved juvenil dermatomyositt (JDM) og utvikles ofte før muskel-manifestasjonene. Symptomene kan minne om kapillar lekkasje syndrom (capillary leak syndrome).
- Nekrotiserende ulcera (vaskulitt sår) signaliserer alvorlig sykdom og kan sees ved anti-MDA5 positivitet (MDA5 syndromet; vennligst se nedenfor) og ved koeksisterende malignitet (9).
- Neglesenger. Hos 30-60 % sees negleseng forandringer med hypertrofi av kutikkelen (mellom neglen og huden) og små hemoragiske infarkter. Kapillaroskopi forventes da å vise patologiske funn.
- Kalsinose ses oftest hos barn og ungdom med dermatomyositt (juvenil dermatomyositt), oftest over knær og albuer der en utsettes for belastning.
- Pannikulitt (fettvevsnekrose), vaskulitt sår og alopeci.
- Lipodystrofi kan utvikles som en komplikasjon til dermatomyositt og tolkes ofte feilaktig som lokalisert muskelatrofi. Andre eksempler på årsaker til lipodystrofi er steroidinjeksjoner, HIV infeksjon, insulinresistens, polycystisk ovarie-syndrom og systemisk lupus erythematosus (SLE).
Dermatomyositt sine dermatitis. Muskelsvakhet, økte muskelenzymer og typiske histologiske dermatomyositt-forandringer uten hud-symptomer forekommer og betegnes sine dermatitis.
Interstitiell lungesykdom (ILD)
Fortetninger i lungevevet er en vanlig ved myositt/dermatomyositt. Det er derfor viktig å undersøke for dette både ved sykdomsdebut og ved senere lungesymptomer. Lungefortetninger påvises ofte når pasienten har anti-MDA5- (avsnitt nedenfor) og antisyntetase antistoff i blodet (vennligst se antisyntetase syndromet i eget kapittel). Totalt er prevalensen av lungemanifestasjoner ved myositt beregnet til ca. 30 % (Lilleker JB, 2017; Tansley SL, 2013).
Anamnese: Vanligste lungesymptom er dyspné ved belastning og tørrhoste.
Lungefunksjonstester viser et restriktivt mønster med redusert gassdiffusjons-kapasitet (DLCO). FVC reduseres også, mens FEV1 i større grad forblir bevart. Dette medfører at en FEV1/FVC ratio >0,7 som tegn på restriktiv lungesykdom (til forskjell fra obstruktiv lungesykdom).
HRCT thoraks viser vanligvis et mønster som ved non-spesifikk interstitiell pneumoni (NSIP), med mattglassfortetninger (ground glass). Mindre vanlig er «usual interstitial pneumonia» (UIP) med utseende av «bikake mønster» (4). UIP er irreversibel og medfører den dårligste prognosen.
Komplikasjoner. Lunge-manifestasjonene kan kompliseres ved pulmonal hypertensjon, sjeldnere pneumo-mediastinum. ILD er sammen med malignitet og infeksjoner vanligste årsak til redusert overlevelse ved myositt (Marie I, 2001; Yamasaki Y, 2011).
Der er beskrevet hjerte-affeksjon ved myositt mellom 9-72 % (Zhang L, 2012). Denne er ikke sjelden subklinisk og omfavner myokarditt, arytmier, hjertesvikt og økt aterosklerose tendens (Schwartz T, 2016).
Artritt forekommer ofte blant pasienter med anti-MDA5 positivitet og antisyntetase syndrom (ASS, se eget kapittel). Ved fravær av anti-CCP antistoff (se revmatoid artritt) er artrittene med myositt vanligvis ikke erosive. Ved ASS med artritt ses anti-CCP hos ca. 1/3 av (Meyer A, 2015).
Tabell. Kjennetegn til hjelp i diagnostikk av ulike myositter. Modifisert etter Leitlinien für Diagnostik und Therapie in der Neurologi, DGN, 2022 (dgn.org/leitlinien)
| Kjennetegn | Polymyositt | Dermatomyositt (DM) og JDM | Inklusjonslegememyositt | Immunmediert myositt | Antisyntetase syndrom |
| Kvinner: menn | 2:2 | 2:1 | 1:3 | kvinner>menn | kvinner>menn |
| Vanligste debutalder | alle aldre | DM: fra 16 år. JDM til 16 år | >45 år | vanligvis >18 år | vanligvis voksen alder |
| Forløp | akutt-subakutt | akutt-subakutt | kronisk >12 måneder | akutt-subakutt >80% | akutt-subakutt |
| Kraftsvikt/pareser | proksimal>distal | Proksimal>distal | Proksimal=distal, asymmetrisk. Kne-strekkere ≥hofte-fleksorer. Finger-fleksorer ≥ skulder-abduktorer. | proksimale underekstremiteter > overekstremiteter. Symmetrisk. | Proksimal >distal |
| CK | opp til 50x | normal til 50x | normal til 15x | opp til 50x | opp til 50x |
| EMG og MR | EMG: myopatisk i aktive faser. MR: Ødem, kontrastmiddel-opptak. Atrofi og fibrose/fettinnlagring i kroniske forløp. | EMG: myopatisk i aktive faser. MR: Ødem og kontrastmiddelopptak. Atrofi og fibrose/fettinnlagring i kroniske forløp. | EMG: myopatisk. I langt kommet stadium ses også kronisk nevrogene forandringer. MR: lite eller ingen aktivitetstegn, men tydelig bindevev/fettinnlagring. | EMG: myopatisk i aktiv fase. MR: Ødem og kontrastmiddelopptak. Ved kronisk forløp ses atrofi og fibrose/fettinnlagring. | EMG: Myopatisk i aktiv fase. MR: Ødem, kontrastmiddelopptak. Ved kronisk forløp ses atrofi og fibrose/fettinnlagring. |
| Dysfagi/ekstramuskulære manifestasjoner: hud, hjerte, lunger | Dysfagi mulig. Ikke hudmanifestasjon. Myokarditt mulig. Interstitiell lungesykdom (ILD) | Dysfagi mulig. Hudaffeksjon er typisk. Myokarditt sjelden. Lungesykdom særlig ved MDA5 antistoff. JDM: kalsinose. | Dysfagi typisk (2/3). Ikke hud-manifestasjon. Sjelden og mild kardial affeksjon. | Dysfagi mulig. Ikke hudmanifestasjon. Myokarditt mulig. Sjelden lungemanifestasjon. | Dysfagi er vanlig initialt. Mekanikerhender, Raynauds fenomen. Artritt. Lungemanifestasjon kan være alvorlig. Kardial manifestasjon mulig. |
| Kreft-assosiasjon | Uvanlig | hos voksne. Særlig ved TIF1γ og NXP-2 antistoff. | nei | Sjelden. HMGCR eller uten antistoff disponerer litt. | Nei |
| ILD | 20-80% | Klinisk 20%, HRCT 78%. MDA5 antistoff, alder >45 år, feber, artritt og høy CRP disponerer for aggressiv ILD | Nei | 4% | 20-80%. Kjernesymptom ved antisyntetase syndromet |
| Muskelbiopsi | Som immunmediert myopati eller uspesifikk myositt. Perivaskulær perimyseal/endomyseal inflammasjon. Endomyseale CD8+T-celler | Perifascikulær atrofi. Perivaskulær inflammasjon. Redusert kapillærtetthet, MHC-1 ekspresjon på perifascikulære fibre. | Inflammatorisk infiltrat, sarkoplasmatiske “rimmed vacuoles”, amyloid-avleiringer, inklusjonslegemer, Ragged red Fibers, COX negative fibre. | Nekrotiske og regenererende fibre, minimale celleinfiltrater | Som ved immunmediert myopati eller uspesifikk myositt. Perivaskulær perimyseal inflammasjon. Endomyseale CD8+ T-celler |
Undersøkelser
Myositter er en sjelden og kompleks gruppe sykdommer. Det kan være vanskelig å skille myositt fra andre, ikke-inflammatoriske muskelsykdommer. Etter anamnese, klinisk undersøkelse og påvisning av høy CK, bør man bestemme hvilke antistoff som kan foreligge. Utenom ANA-test er myositt-spesifikk “myositt-blott” aktuelt. Disse som tilbys av immunologiske laboratorier (Ashton C, 2021).
Anamnese: Familie anamnese. Hvilke symptomer dominerer: svakhet, smerte eller stivhet? Når oppstår symptomene; I hvile, under eller etter fysisk anstrengelse? Debut alder? Hvor raskt har symptomene utviklet seg? Medikament forbruk (statiner, andre)? Symptomer på andre systemiske bindevevssykdommer? Foreligger symptomer fra lunger, hud eller andre aktuelle organer?
Klinisk undersøkelse: Det gjøres helst en generell undersøkelse som omfatter lunger, hjerte og andre organer. Muskulaturen vurderes spesielt; proksimal / distal, symmetrisk / asymmetrisk? Hypertrofi / atrofi? (Hypertrofi ses hos noen pasienter med muskeldystrofi). Fascikulasjoner? (Ses ved nevromuskulære tilstander).
-6-minutter gangtest gir en vurdering av fysisk gangfunksjon (antall meter tilbakelagt i løpet av 6 minutter rask gange). Ved kontroller over tid kan eventuell sykdomsprogresjon synliggjøres.
-Test av muskelstyrke. Ved orienterende undersøkelse kan musklenes form og størrelse avdekke hypertrofi, atrofi, asymmetri, fascikulasjoner eller spenninger. Smerter ved palpasjon/lett trykk vurderes, samt tegn til kraftsvikt når pasienten reiser seg fra stolen og fra huk uten å støtte seg. Tilsvarende bør armene kunne eleveres over horisontalt nivå uten problemer og mot lett motstand. Standardiserte tester er viktige verktøy i oppfølging av pasienter, særlig for å vurdere behandlingseffekt og om behandlingsmål oppnås. Fysioterapeuter tester både direkte muskelstyrke og utholdenhet (Oldroyd AGS, 2020). Til test av myositt pasienter er der utviklet to tester; Muskelstyrke ad modum Kendall og FI2 test (Alexanderson H, 2006; Kendall HO, Kendall FP, Wadsworth GE. Muscles : testing and function. 2nd ed. ed. Baltimore: Williams and Wilkins; 1971).
Laboratorieprøver. Generelt kan prøvene omfatte CRP, SR, Hb, leukocytter med differensialtellinger, elektrolytter, lever-, nyre- og thyreoidea-funksjonsprøver, kreatin kinase (CK), LD og glukose. ved mistanke om kardial manifestasjon kan troponin være aktuelt. Vær oppmerksom på at Troponin T kan øke ved myositt, også uten sikker hjertemuskelaffeksjon. Cortisol (morgenverdi) er aktuelt ved mistanke om binyre over- eller under-funksjon. Urin stiks, ved høy CK, kan myoglobin i urin være aktuelt.
-Muskelenzymer; CK, LD, ASAT. Disse er vanligvis forhøyet, men ikke alltid. Noen ganger finner man normal CK med moderat forhøyet LD. CK stammer fra muskelcellenes cytoplasma (sarkoplasma), og mesteparten utgjøres av isoenzymet CK MM. Nivået av CK ligger oftest mellom 2-20 ganger øvre normalverdi. CK- forhøyelse sees også ved mange andre tilstander som traume, eksessivt muskelarbeid, trening (overtrening: rabdomyolyse), infeksjoner, toksisk påvirkning og de non-inflammatoriske muskeldystrofiene. Hos ca. 0,7% friske er CK idiopatisk forhøyet (Lilleng H, 2011). Forhøyede CK -nivåer halveres ca. hver 24-36 timer dersom utløsende årsak er fjernet. Normalisering i løpet av 3-5 dager forventes, slik at noen dagers treningspause før prøvetakning anbefales. -Troponin I, troponin T; Troponin I er en mer følsom markør for hjerteaffeksjon enn Troponin T. -ANA, myosittspesifikke- og assosierte antistoffer; vennligst se immunologiske undersøkelser nedenfor:
Immunologiske undersøkelser. Myositt spesifikke antistoffer (MSA) er immunologiske tester som nesten utelukkende sees ved inflammatoriske myopatier. Dette til forskjell fra myositt-assosierte auto-antistoffer (MSA) som sees også ved andre systemiske bindevevssykdommer. MSA har de siste 20-årene bidratt betydelig til diagnostikk, utredning og behandling av myositt.
Tabell 1. Oversikt over myositt spesifikke antistoff (modifisert etter Lundberg I, 2021)
| Antistoff | Forekomst | Kliniske kjennetegn |
| Antisyntetase-antistoff | Antisyntetase syndrom; Proksimal myositt (PM eller DM), ILD, artritt, Raynauds, mekaniker hender og feber, Utslett på halsen. | |
| -Anti-Jo-1 | 20% | Antisyntetase-syndrom |
| -Anti-PL-7 | 5-10% | Antisyntetase-syndrom |
| -Anti-PL-12 | <5% | Antisyntetase-syndrom |
| -Anti-EJ | 5-10% | Antisyntetase-syndrom |
| -Anti OJ | <5% | Antisyntetase-syndrom |
| -Anti KS | <5% | Antisyntetase-syndrom |
| -Anti-Zo | <1% | Antisyntetase-syndrom |
| -Anti YRS | <1% | Antisyntetase-syndrom |
| Immunmediert myopati (IMNM) | ||
| -Anti-SRP | 5-10% | Nekrotiserende myopati, høy CK, «hissig», hjerte |
| -Anti-HmG-CoA reduktase | 5-10% | Nekrotiserende myopati med høy CK, statin bruk |
| Dermatomyositt | ||
| Anti-Mi2 | 5-10% blant dermatomyositt (DM) | Typiske hudforandringer: panne og rundt øyne (heliotropt), sjal-tegn, V-tegn, Gottrons tegn og papler, Proksimal myositt. |
| Anti-TIF1gamma | 20% blant DM | Assosiert med malignitet. Klinisk manifestasjon: skalp eksem, periorbitalt ødem, heliotropt utslett, poikoderma (pigmentforandringer), ulcera og nekroser, kløe, lipodystrofi, dysfagi, myositt, Gottrons tegn og papler. Proksimal myositt. |
| Anti-NXP2 | 25 (hos DM og Juvenil DM) | Assosiert med malignitet. Hos JDM; assosiert med kalsinose |
| Anti-MDA5 | Amyopatisk (50%) DM; Alvorlig ILD, ulcerasjoner, alopeci, periorbitalt ødem, Gottrons tegn og papler, mekaniker hender, negleaffeksjon, pannikulitt. (MDA5-syndromet) | |
| Anti-SAE | 5% blant DM | Hissig hudaffeksjon, dysfagi. Ikke relatert til malignitet |
| Inklusjonslegememyositt | ||
| Anti-cytosolic 5′-nucleotidase 1A (5NT1A) | Sensitivitet 30- 50% (referanse: Felice KJ, 2018), dessverre ikke helt spesifikt for IBM og ses også ved andre tilstander | Dysfagi, myositt kan være asymmetrisk. Distal myositt og atrofi ved håndledd og fingerfleksorer, kne-ekstensorer og ankel-dorsalfleksorer. |
| Overlapp-syndromer | ||
| Ku, RNP, Ro/SSA, La/SSB, PmScl | Overlapp mot andre systemiske bindevevssykdommer | Ku: RA, Systemisk sklerose, myositt og overlappsyndromer. RNP: MCTD. Ro og La: Sjøgrens, subakutt kutan lupus. PmScl: systemisk sklerose |
Bildediagnostikk
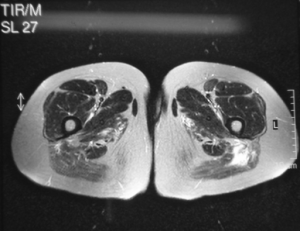
MR-undersøkelse.
- Lårmuskulaturen undersøkes oftest, da det er størst sjanse for å se forandringer her.
- Muskel- og fascieødem tyder på inflammasjon, mens fettinfiltrasjon og atrofi tyder på kroniske forandringer.
- MR-bilder kan til en viss grad differensiere mellom poly- og dermatomyositt. Dermatomyositt er ofte preget av mer fascieødem (Pilania K, 2021).
- Ødem og fett-infiltrasjon er ikke spesifikt for myositt og kan sees også ved andre tilstander, for eksempel muskeldystrofier.
- MR av muskulatur brukes også til å å finne egnet sted for biopsi, da muskelinflammasjonen ofte er flekkvis fordelt.
Ultralyd med Doppler kan i noen tilfeller identifisere områder med muskelinflammasjon. Undersøkelsen er da nyttig for lokalisering av muskelbiopsi og for vurdering av behandlingsrespons (Conticini E, 2023).
Kapillaroskopi av neglefolder kan vise patologiske forandringer med megakapillærer og endret arkitektur. Dette kan minne om systemisk sklerose, spesielt ved tilstedeværelse av antistoffene MDA5 eller TIF-1-γ foreligger (Mugii N, 2023).
CT-undersøkelser. HRCT lunger tas for å utelukke lungeaffeksjon. Konvensjonell CT-undersøkelse brukes ved mistanke om malignitet. Ved lunge-manifestasjon viser HRCT initialt et NSIP mønster, oftest lokalisert dorso-basalt bilateralt. Senere i forløpet kan fibrose og et UIP mønster forekomme. Utseende og lokalisering av lungevevsaffeksjonen kan gi indikasjoner på hvilken subtype myositt eller antistoffgruppe (antisyntetase, anti-MDA5, anti-SRP) pasienten tilhører (Laporte A, 2022). Vennligst les om lungesykdom i eget kapittel.
Røntgen øsofagus med svelging av barium kontrastmiddel kartlegger dysfunksjon ved svelging. Affeksjon av svelgemuskulatur (dysmotilitet) er ikke uvanlig ved myositt og er et dårlig prognostisk tegn. Det kan også være assosiert med aggressiv myositt og med malignitet (Lu X, 2014). Dysmotilitet og svelgevansker kan øke risikoen for aspirasjon og pneumoni. De kan også kan på sikt medføre alvorlig vekttap og behov for sonde- eller PEG-ernæring.
PET/CT kan vurderes for utelukke tegn til malignitet, særlig ved dermatomyositt hos voksne. Undersøkelsen kan også vise eventuell inflammasjon i lunger (Yildiz H, 2022).
Elektromyografi (EMG) kan skille mellom nevrogen (non-inflammatorisk) og inflammatorisk muskulær (myositt) skade. Blant annet måles insersjonsaktivitet, som er elektrisk aktivitet utløst av nålestikket. Dette er redusert ved muskelcelletap og økt ved denervasjons-myopati, inflammasjon og metabolske prosesser. Abnorm spontan depolarisering er alltid et tegn på patologi. Ved myositt ses polyfasiske motor unit potentials (MUP), «sharp waves» og repetitive utladinger. Bruk av EMG i utredningen har avtatt og ofte erstattet av de andre metodene (Meyer H-J, 2018).
EKG er aktuelt ved mulig hjerte-manifestasjon.
Muskelbiopsi
. CC BY-NC-SA 3.0")
Biopsi kan vise inflammasjon og nekrose, og kan dermed skille mellom polymyositt, dermatomyositt og inklusjonslegeme myositt.
-Perimyseal inflammasjon. Dermatomyositt (DM) kjennetegnes av perimyseal inflammasjon (se foto) med hovedsakelig CD4+ lymfocytter mellom celler og muskelfascikler. Perifasciculær atrofi (kapillærtap og ischemisk skade), redusert kapillartetthet og “membrane-attack-complex” deponering er typiske funn ved DM.
-Endomyseal inflammasjon. Polymyositt (PM) har endomyseal inflammasjon med opphopning av lymfocytter som omgir og invaderer non-nekrotiske fibre, hovedsakelig CD8+ cytotoksiske celler.
-Ved antisyntetase syndrom (ASS) finner man deponering av C5b-9 komplekser og inklusjoner av myofilament. Forandringene er lokalisert perifascikulært, som ved DM (Stenzel W, 2015). ASS er beskrevet i eget kapittel.
. CC BY 4.0")
-Inklusjonslegeme myositt (IBM) har perimyseal inflammasjon med CD4+ lymfocytter, i tillegg til ragged red fibers som tyder på mitokondrieskade. Filamenter opptrer både i cytoplasma og i nukleus, og sees som inklusjoner i mikroskop. Ved elektronmikroskopisk finner man mikrotubulære filamenter som ved farging med Kongo rødt indikerer Beta amyloid protein. Det sees også vakuoler med amyloid innhold (rimmed vacuoles= vakuoler som er begrenset av en tykk vegg). IBM er også omtalt i eget kapittel
–Immunmediert nekrotiserende myopati (IMNM) har typisk histologi med uttalt nekrose og lite lymfocyttinfiltrasjon. Biopsi-svar ligner da rabdomyolyse og toksisk myositt.
Lungefunksjonstester. Opptil 2/3 av myositt-pasientene har lungeaffeksjon med interstitielle forandringer. Symptomene kan være subkliniske, og det er derfor viktig å vurdere lungefunksjon med tanke på progresjon. Lungefunksjons-tester med vurdering av FVC og FEV og gassdiffusjon (DLCO), gir en tilstrekkelig vurdering av lungefunksjonen. HRCT thoraks (se ovenfor) bør utføres initialt og ved mistanke om progresjon. Vennligst les om lungefunksjonstester i eget kapittel
Malignitetsutredning
Myositt, spesielt dermatomyositt hos voksne, har økt risiko for underliggende kreftsykdom. Malignitetsutredning er derfor indisert. Anbefalt utredning inkluderer:
- CT thorax/abdomen/bekken, alternativt PET-CT
- Mammografi og gynekologisk undersøkelse
- Hemofec
- Ev. benmargsbiopsi og mage-tarm endoskopi
De vanligste tumorene assosiert med myositt er ovarial-, bryst-, lunge-, tarmkreft og lymfom (Dobloug GC, 2015). Vennligst les om malignitet ved revmatiske sykdommer i eget kapittel.
Utredningen av myositt er ofte komplisert og krever samarbeid mellom spesialister som revmatolog, nevrolog, endokrinolog, spesialist i biokjemi og patolog. I praksis er det oftest revmatologer som tar ansvar for oppfølging av de inflammatoriske myosittene, mens nevrologer tar ansvar for non- inflammatoriske myopatiene (muskeldystrofier med flere).
Diagnostiske kriterier/klassifikasjons kriterier
I 1975 publiserte Bohan og Peter (Bohan A, 1975) diagnostiske kriterier for myositt;
*Forutsetter at andre årsaker til myopati er ekskludert
- 1. Symmetrisk proksimal muskelsvakhet
- 2. Forhøyde muskelenzymer i serum (CK, aldolase, ASAT, ALAT, LD)
- 3. EMG med polyfasiske MUP, ”sharp waves” og repetitive utladninger
- 4. Muskelbiopsi; Degenerasjon, regenerasjon, nekrose, fagocytose og interstitielle mononukleære infiltrater
- 5. Typiske eksantem (som heliotropt eksantem, Gottrons tegn/papler)
- For å stille diagnosen DM må kriterium 5 være til stede
- Mulig PM/DM = 2 kriterier
- Trolig PM/DM = 3 kriterier
- Definitiv PM/DM = 4 kriterier
* Kriteriene forutsetter at andre årsaker til myopati er ekskludert.
Klassifikasjonskriterier EULAR/ACR 2017
Bohan og Peter kriteriene brukes til viss del fortsatt, men der det i 2017 ble publisert nye klassifikasjons kriterier (Lundberg IE, 2017).
Kriteriene deles opp i om det foreligger muskelbiopsi eller ikke. Poeng score på >9 viser høy sannsynlighet for myositt og kriteriene går deretter videre for å klassifisere undergrupper;
Kriteriene summerer score som bygger på følgende parametere med eller uten muskelbiopsi tilgjengelig
Kalkulator for EULAR/ACR kriterier, vennligst se her
Med disse kriteriene oppnås en sensitivitet og spesifisitet på 90 %.
Spesielle myositt-undergrupper
Antisyntetase syndrom, ASS
Alle myositter med antisyntetase antistoffer (oftest anti-Jo-1, anti-PL7 eller anti-PL12) har høy forekomst av interstitiell lungesykdom. Opptil 30% av alle med polymyositt eller dermatomyositt kan defineres som ASS (Klassisk triade med ILD, myositt og ofte artritt, kan utvikles over tid. I tillegg har mange Raynauds fenomen, mekanikerhender (sprekkdannelse på fingre) og feber (Cavagna L, 2015). Vennligst les om ASS i eget kapittel.
Orbital/okulær myositt

Øye-manifestasjon i form av orbital myositt kan gi periorbital smerte, diplopi og hevelse av øyelokk. Diagnose ved MR-undersøkelse og biopsi. Oppfølging hos øyelege.
Immunmediert nekrotiserende myopati (IMNM)
Pasienter med anti-HmGCoA-reduktase (HMGCR) eller SRP -antistoffer: Disse pasientene har ofte immunmediert nekrotiserende myopati (IMNM), en “hissig” form for myositt. I løpet av få uker kan IMNM føre til betydelig muskelsvakhet, inkludert dysfagi og høy CK. Hjerteaffeksjon kan forekomme, spesielt ved anti-SRP, men lunge-manifestasjoner er sjeldnere. Histologisk sett viser IMNM nekrose uten inflammatoriske infiltrater, et bilde som kan ligne cancer-assosiert myopati (Pinal-Fernandez I, 2018).
-
Anti-HmGCoA-reduktase-relatert myositt: Denne typen myositt er assosiert med statinbruk (kolesterolsenkende medikamenter), men kan også forekomme hos ca. 50% uten statinbruk.
- Seronegative IMNM og HMGCR myopatier: Disse pasientene har en mulig økt forekomst av kreft, og screening kan være aktuelt. (Yang H, 2017).
Behandling:
- Anti-SRP positiv myositt: steroider og rituksimab.
- Anti-HmGCoA-reduktase myositt: IVIG i monoterapi.
Vennligst les også om statin-myopati i eget kapittel.
Kreft-assosiert myositt
Når myositt oppstår kort tid før eller etter kreftsykdom, er det ofte en sammenheng. Mistanken om kreft styrkes ved tilstedeværelse av anti-TIF1γ og anti-NXP2 antistoff, som ofte sees ved dermatomyositt (Fiorentino DF, 2013). Anti-NXP2 antistoffer kan også påvises hos barn med juvenil dermatomyositt uten å være kreft-assosiert, men er da vanligvis assosiert til kalsinose. Som nevnt ovenfor kan seronegative IMNM og HMGCR myopatier også medføre en viss økt risiko for kreft (Yang H, 2017).
Dermatomyositt har økt insidens av malignitet når den oppstår hos voksne. Det er ikke uvanlig at relatert kreft oppdages tre år før og etter debut av dermatomyositt symptomer (Dobloug GC, 2016). Internasjonale retningslinjer anbefaler årlig malignitet-screening i 3-5 år etter DM-diagnose hos voksne pasienter. Andre risikofaktorer for malignitet er høy alder, mannlig kjønn og dysfagi (Lu X, 2014). Bryst-, gynekologisk-, GI- og lungecancer er de vanligste typene kreft ved kreft-assosiert myositt (Yang, 2017).
En viss utredning for kreft kan gjøres ved påvist myositt/dermatomyositt. Se ovenfor og kapittel om kreft ved revmatisk sykdom.
Anti-MDA5 myositt

Pasienter med anti-MDA5 antistoff har mindre aktiv myositt sammenlignet med andre typer myositt. De kan imidlertid ha betydelig hud-affeksjon, inkludert nekrotiske sår. Flere av pasientene utvikler «rapid progressive lung disease», en raskt progredierende lungesykdom som kan være meget alvorlig og vanskelig å behandle. Utvikling av pneumo-mediastinum er ikke uvanlig og er et dårlig prognostisk tegn.
Behandlingsalternativer i tillegg til steroider er rituksimab, cyclofosfamid, takrolimus, plasmaferese og intravenøse immunglobuliner (Mehta P, 2021).
Inklusjonslegeme myositt
Inklusjonslegeme myositt (IBM) skiller seg mest ut fra de andre myosittene. Den er dobbelt så hyppig hos menn og debuterer vanligvis etter 50 års alder. IBM har en langsommere progresjon sammenlignet med andre myositter. Pasienter med IBM kan ha affeksjon av distal muskulatur (finger-fleksorer, tibialis anterior) og asymmetrisk fordeling. Mange har affeksjon av svelgmuskulatur, men ikke interstitiell lungesykdom (ILD). IBM svarer dessverre dårlig på immunsuppressiv behandling. IBM er nærmere beskrevet i eget kapittel.
Infeksjons-indusert myositt.
Virus
Hepatitt E er endemisk i Asia og Afrika, men sjelden i vår del av verden. Vanligvis er sykdomsbildet mildt, men tilfeller med alvorlig myositt er sett (Mengel AE, 2015). Også nevrogene smerter kan være en del av sykdomsbildet (Abravanel F, 2018). Inkubasjonstid er vanligvis 4-6 uker (Folkehelseinstituttet, 2019; Waquar S, 2021).
Andre. Hepatitt B og C, COVID-19, parvovirus, CMV med flere kan en sjelden gang indusere myositt (Narayanappa G, 2021; Saud A, 2021), men også polymyalgia revmatika-lignende sykdombilde (Ottavani S, 2022).
Bakterier, sopp og parasitter
Stafylokokker- og en rekke andre bakterier, sopp og parasitter kan medføre myositt, men er sjelden (Narayanappa G, 2021).
Svangerskap
Det er rapportert økt forekomst av spontanaborter, dødfødsler og for tidlige fødsler ved myositt. Pasienter med aktiv myositt er mest utsatt. På grunn av muskelsvakhet, kan forberedelse til fødsel ved sectio være aktuelt (Ito Y, 2021). Vennligst se også info fra NKSR.
Differensialdiagnoser
Mange tilstander kan etterligne myositt, men en grundig vurdering (se “Undersøkelser” ovenfor) ovenfor vil vanligvis avgrense disse.
Hyppige differensialdiagnoser:
- Kroniske generaliserte muskelsmerter ved fibromyalgi: Ofte sett hos kvinner.
- Polymyalgia revmatika: Kan oppstå hos eldre personer (over 70 år) i løpet av dager.
- Hypothyreose: Lavt stoffskifte.
- Tyreotoxikose: Høyt stoffskifte.
- Steroid-myopati: Kan oppstå ved langvarig bruk av kortikosteroider, særlig i høye doser.
En stor gruppe sjeldne tilstander som kan kreve nærmere differensialdiagnostisk utredning ved “seronegativ” eller atypisk myositt er non-inflammatorisk myopati med late-onset muskeldystrofier og metabolske myopatier, samt Lambert-Eaton myasteni (ved malignitet), mitokondrie-myopatier, diabetisk myonekrose og infeksiøse myopatier (Harmann P, 2013). Immunmediert nekrotiserende myopati kan histologisk være vanskelig å skille fra rabdomyolyse eller toksisk myopati av annen årsak fordi det er lite eller ingen inflammasjonstegn i muskelcellene. Imidlertid vil mange ha antistoff i form av anti-HMGCR eller anti SRP.
Generelt bør distal muskelsykdom, slik som manifestasjoner i finger-fleksorer og fot-ekstensorer eller fascikulasjoner tilsi utredning for inklusjonslegeme myositt, men også nevromuskulær sykdom som amyotrofisk lateralsklerose (ALS) og Charcot-Marie-Tooth sykdom.
Liste over differensialdiagnoser:
- ALS (amyotrofisk lateralskerose). Distal affeksjon, hyperrefleksi, muskel fibrillasjon, spasmer, lett økt CK
- Amyloid myopati. Subkutane forandringer, proteiner/eggehvite i urinen. Typisk biopsi (farging med Kongorødt).
- Carnitine-palmitoyl-transferase II mangel; Muskelsmerter
- Diabetisk muskelsvinn (amyotrofi) og diabetisk muskelinfarkt: Diabetes Type 2 med akutt, asymmetrisk start med fokal smerte og svakhet i lår, ofte vekttap. EMG skiller fra myositt
- Elektrolytt mangel: Hypokalemi, hypokalsemi, hypomagnesemi
- Eosinofil myositt: Differensialdiagnoser er Hypereosinofilt syndrom: IgE, eosinofile leukocytter > 1.5 × 109/L, hudaffeksjon. Multiorganaffeksjon, smerter og svakhet i proksimal muskulatur, Raynauds, vaskulitt i små blodårer, CK kan være forhøyet
- Eosinofil fasciitt (CK oftest normal, typiske funn i hud og underhud)-
- Fibromyalgi. Vanlig blant ellers frisk kvinner, generelle smerter (kronisk i begge over- og underekstremiteter, nakke eller rygg), «Tenderpoints», normale prøver.
- Glykogen lagringssykdom type V: McArdles sykdom/ fosforylase mangel. Smerter/kramper etter aktivitet. Glykogen lagringssykdom type II, Adult acid maltase mangel (Pompes sykdom; Infantil, meget alvorlig form og adult, «late-onset» form med gjennomsnittsalder på 36 år ved diagnose, progredierende krafttap, med pustevansker (svake respirasjonsmuskler).
- GVHD (graft versus host) myositt etter transplantasjon. Oftest mer enn ett år etter transplantasjon. Funn som ved myositt.
- HIV infeksjon. Kronisk vekt- og krafttap, klinisk bilde med CK stigning, (inflammatorisk myopati) som ved myositt. HIV-test. CD4 T-celler korrelerer dårlig med myopati-symptomer.
- Hypothyreose: Høy TSH, Lav f-T4.
- Inklusjonslegeme myositt; Lavere kreatin kinase (CK) i blod, svært langsom progresjon, høyere alder, distal affeksjon, ingen antistoff, typisk biopsi.
- IMNM (immun-mediert Nekrotiserende Myositt). Kan være relatert til statiner (kolesterolsenkende medikamenter). HMGCR antistoff foreligger hos noen.
- Kalsinose (kalkansamlinger under huden og i bløtdelsvev) forekommer ved systemisk sklerose, juvenil dermatomyositt og ulike tilstander.
- Katapleksi. Plutselig tap av muskelkraft i flere anfall. Ved narkolepisi har 70% også katapleksi.
- Kennedy sykdom (Spinal and bulbar muscular atrophy, SBMA): Svekket kraft og symmetrisk muskelatrofi i proksimale muskler. Kreatin kinase (CK) er moderat forhøyet. Menn i 40-50 års alder. Redusert androgen sensitivitet med gynekomasti, testes atrofi og redusert libido. Fascikulasjoner i ansiktsmuskulatur. Tale- og svelgevansker. Skilles fra myositt ved klinisk bilde (ansiktsmuskler, gynekomasti, EMG og muskelbiopsi).
- Lambert-Eaton syndrom: Myasteni-lignende, kreft-relatert (paraneoplastisk syndrom), ikke øyemuskel affeksjon, antistoff mot voltage-gated calsium kanal (VGCC), EMG funn.
- Makro-CK. Makroenzymer er normale enzymer (eller isoenzymer) som binder seg enten til immunglobuliner (IgG) = Type 1 eller lipoproteiner og andre substanser (Type 2) og dermed akkumuleres i serum. De er ikke frie enzymer, men forårsaker falsk forhøyede CK-målinger. Makro-CK ses oftest hos personer over 60 år. Makro-CK kan foreligge med- eller uten assosiert sykdom, inklusiv kronisk leversykdom og malignitet.
- Medikamentindusert myopati. Statiner. Smerter med eller uten forhøyet kreatin kinase (CK) i blod. Medikamentanamnese viktig. Høye kortikosteroid-doser (Prednisolon) over lang tid: Steroid myopati: CK normal. Andre: Amiodaronhydroklorid (Cordarone) mot hjerterytme forstyrrelser, Plaquenil, kokain, alkohol, kolkisin.
- Mitokondrie myopati, encefalomyopati, laktat acidose, slag-lignende symptomer (MELAS)
- Muskeldystrofi (arvelige, progressive muskelsykdommer). Bør alltid overveies ved kronisk muskelsykdom.
- Myastenia gravis: Økt fysisk muskulær trettbarhet, ofte i ansiktsmuskler, inklusiv øyne, normal CK, EMG er karakteristisk, anti-acetylkolin antistoffer
- Myoklonisk Epilepsi med «Ragged Red Fibers» (MERRF). Mitokondriesykdom. “Ragged Red Fibers” er sammeklumpede syke mitokondrier i vevsprøve (biopsi), lav kroppshøyde, hørselstap, laktat acidose, intoleranse for fysisk trening.
- Parkinsons sykdom. Tremor, rigor, påfallende langsom gange.
- Periodisk paralyse
- Polymyalgia revmatika. Brå start, uttalt morgenstiv, smerter og stivhet, ikke primært muskelsvak, høyere alder, høy SR og CRP. Normal CK
- Post polio syndrom. 15-30 år etter poliomyelitt (35-60 års alder). Muskelsmerter, Svakhet, Utmattelse
- Rabdomyolyse: CK er svært høy over 10.000 (Obs! nyreskaderisiko). Etter skade, intoksikasjon eller kritisk sykdom. Rabdomyolyse og myalgi syndrom assosiert med RY1 mutasjon. Økt risiko for hypertermi. Kan ha moderat forhøyet CK og muskel-manifestasjoner
- Refsums sykdom. Retinitis pigmentosa, kronisk polynevropati med pareser, muskelatrofi distalt, cerebellære symptomer med ataksi.
- Sarkoid myopati: Oftest asymptomatisk, sarkoidose i andre organer, granulomer i biopsi.
- Skleromyositt (sykdom med overlappende symptomer mot systemisk sklerose)
- Spinal muskelatrofi: Hypertrofi av legger (Ørstavik K, 2020)
- Statinmyopati
- Steroid-myopati; Muskelsvakhet ved langvarig kortikosteroider. Normal CK
- Subaraknoidal-blødning og slag
- Trichionose: Parasitt infeksjon (trikiner) fra inntak av rått (svine)kjøtt. Sterke muskelsmerter og enzym- (CK) stigninger. Eosinofili. Muskelbiopsi bekrefter diagnosen.
- Viral myositt (sjelden) ved: Influensa A and B, H1N1 virus, Coxsackie-virus, Epstein-Barr-virus, herpes simpleks-virus, parainfluensa, adeno-virus, echovirus, cytomegalo-virus (CMV), meslinger-virus, varicella zoster, Human immunodeficiency virus (HIV), Dengue feber. Rask sykdomsutvikling, kan være begrenset til få muskelgrupper. Effekt av antiviral behandling.
Behandling
Behandling er individuelt tilpasset styres av flere faktorer:
- Grad av muskelaffeksjon
- Hvilke organ som er affisert
- Myositt spesifikke antistoff
- Høy alder og ev komorbiditet
- Myositt type
Utenom medikamentell behandling har individuelt tilpasset fysioterapi med opptrening og styrking av muskulaturen vist seg nyttig. Fysisk trening kan bedre mitokondrie-funksjon, angiogenese, muskelvekst og redusere inflammasjon. Aktuelle øvelser tilpasses individuelt og kam omfatte trening på sykkel, moderat aktiv gange fem dager i uken og tilpasset styrketrening av angrepet muskulatur (Alexanderson H, 2018; van Thillo A, 2019).
Steroider med Prednisolon. Dosering er individuell og tilpasset sykdommens alvorlighetsgrad. En dose på 0,75mg-1mg/kg med nedtrapping har vært vanlig førstehånds valg som alvorlig myositt behandling. Ved alvorlig lungemanifestasjon med rask progresjon (spesielt ved antisyntetase syndrom) velges alternativt metylprednisolon intravenøst de første tre dager.
DMARDS. Samtidig behandling med steroider og steroidsparende middel (DMARDs) som metotreksat ev azathioprin eller mykofenolat er vanlig. Ved alvorlig lungemanifestasjon er cyclofosfamid iv et aktuelt alternativ til rituksimab (se biologisk nedenfor) (Maher TM, 2023).
Biologisk. En randomisert placebokontrollert studie med biologisk legemiddel i form av rituksimab hos behandlingsrefraktære myosittpasienter viste at 83 % av pasientene nådde «definition of improvement» (Oddis CV, 2013). Rituksimab ser også ut til å være effektiv ved myositt assosiert ILD (Andersson H, 2015).
Immunglobulin (IVIG 2 g/kg/mnd.). Intravenøs immunglobulin har vist effekt mot polymyositt og dermatomyositt, og velges i ellers behandlingsrefraktære tilfeller, alternativt også ved alvorlig organmanifestasjon og en ugunstig antistoff-profil, slik som en kan se ved alvorlig dysfagi, betydelig organ-manifestasjoner og ved SRP og HMGCR-antistoff (Lundberg IE, 2021; Hoa SA, 2007 Allenbach Y, 2018), blant eldre pasienter eller i svangerskap. I noen tilfeller kombinerer IVIG med immunsuppressiv behandling som azathioprin, metotreksat, mykofenolat (MMF), takrolimus, rituksimab eller cyklofosfamid. IVIG har vist effekt i en randomisert, placebokontrollert studie ved dermatomyositt. Blant mulige bivirkninger er infusjonsreaksjoner og økt tromboemboli-risiko (Aggarwal R, 2022).
Nintedanib (antifibrotisk medikasjon) er vist å kunne redusere progresjon av fibrotisk lungesykdom (Flaherty KR, 2019). I myositt-tilfeller der slik lungemanifestasjon foreligger, kan supplerende behandling med nintedanib vurderes. Et samarbeid med lungelege er da også viktig.
Pneumocystis-profylakse med trimetoprim + sulfonamid (Bactrim) er aktuelt ved sterk immunsuppressiv medikasjon, særlig ved samtidig lungesykdom (Liu L, 2023).
CAR-T cellebehandling er prøvd ut (utprøvende behandling) i få, svært alvorlige, behandlingsrefraktære tilfeller og med lovende resultater (Müller F, 2023; Lundberg IE, 2023; Pecher AC, 2023).
Retningslinjer, anbefalinger og prosedyrer
Lundberg IE, 2017: EULAR Klassifikasjon av myositt
Allenbach Y, 2018; ENMC anbefalinger for IMNM
Revmatologisk forening/Legeforeningen 2020
Prognose
1/3 del av myositt pasientene har monofasisk sykdomsforløp, 1/3 har prolyfasisk sykdomsforløp og den siste 1/3 del har et kronisk forløp. Overlevelse er avhengig av alder, diagnostisk «delay », ev malignitet, og ILD. 5-års overlevelse er ca. 95%, 10-års overlevelse 90 % (Danieli MG, 2014).
I listen nedenfor er lenker til kapitler der andre myopatier er beskrevet:
- Antisyntetase syndrom
- Inklusjonslegeme myositt (IBM)
- Non-inflammatoriske myopatier ( Infeksiøs myositt, toksisk myopati, sekundær myopati til systemisk sykdom, muskeldystrofier. kongenitale myopatier, metabolske myopatier, mitokondrie myopatier. Idiopatiske myopatier.
- Juvenil dermatomyositt
Litteratur
- Sarwar A, 2022 (polymyositt)
- Qudsiya Z, 2021 (dermatomyositt)
- Halilu F, 2022 (antistoff ved myositt)
- Cheeti A, 2021 (autoimmune myopatier)
- Schmidt J, 2018 (diagnose og behandling)
- McGrath ER, 2018(diagnose og behandling)
- Malik A, 2016 (diagnose og behandling)
- Betterige Z, 2015 (myositt-spesifikke antistoff)
