BARN MED REVMATISK SYKDOM. BARNEREVMATOLOGI (REV 053-062)
122 Juvenil Idiopatisk Artritt, JIA, barneleddgikt (REV 054)
Barneleddgikt-Juvenil idiopatisk artritt (JIA)
Jan Tore Gran and Øyvind Palm
Kjennetegn på juvenil artritt (JIA)
Mistenkes ved stivhet, halting eller leddhevelser uten at infeksjon, skade eller annen årsak påvises blant barn som er under 16 år.
Oligo-/pauciartikulær form angriper fire eller færre ledd, polyartikulær form minst fem ledd, entesitt-relatert har sene- og ryggmanifestasjoner og systemisk form kjennetegnes ved feberepisoder og eksantem.
Blodprøver kan vise økt CRP og SR. ANA ved oligoartikulær type, CCP antistoff ved “voksen type” polyartikulær JIA. HLA-B27 ved entesitt-relatert form og høy ferritin ves systemisk type.
ICD10: M08.0–
Prosedyrekoder: Leddpunksjon/artrocentese (klikk for å spesifisere ledd:) TN_10. UL veiledet leddpunksjon: NXA10K. Mikroskopi av leddvæske: NXFT05.
ATC koder (for legemiddelstatistikk): Prednisolon: H02A B06 Immunsuppressive legemidler: L04A A. etanercept: L04AB01, adalimumab: L04AB04, triamcinolon (Lederspan): H02AB08, betametason (Celeston Chronodose): H02AB04, rituksimab: L01XC02, abatacept: L04AA24, tocilizumab: L04AC07, infliksimab: L04AB02.
Definisjon av JIA
Juvenil Idiopatisk Artritt (JIA, barneleddgikt) ble tidligere kalt juvenil revmatoid artritt (JRA) og juvenil kornisk artritt (JCA). JIA er en heterogen gruppe inflammatoriske sykdommer i ledd og rygg som debuterer blant barn før 16 års alder (Garner AJ, 2021). Basert på klinisk forløp og til dels på antistoffer og genetikk, kan sykdommen deles inn i seks hovedgrupper (Tatayatikom A, 2023):
1) Oligoartikulær artritt (inntil 4 angrepne ledd første 6 måneder) som er vanligst, særlig blant jenter. Utslag i ANA-test i blodet er ofte tilstede.
2) Polyartikulær artritt (5 eller flere artritt-ledd første 6 måneder), noen har utslag i revmafaktorer (RF, a-CCP antistoff: “seropositive”)
3) Entesitt-relatert artritt som angriper store ledd, senefester (enteser) og columna med iliosakralledd, oftest blant gutter som genetisk er HLAB-27+
4) Juvenil psoriasisartritt der psoriasis i nærmeste familie er vanlig.
5) Systemisk JIA (Stills sykdom) som kjennetegnes ved høy feber og i blodet er ferritin høy.
6) Udifferensiert type passer inn i de andre gruppene eller overlapper.
Historie
De første beskrivelsene av kronisk artritt hos barn ble gjort i 1864 (Cornil MV, 1864 Compte rendu Med Soc Bio Series, Paris). Før dette skilte en ikke sykdommen fra revmatisk feber. Til å begynne med ble sykdommen ofte oppfattet å være identisk med artritt hos voksne, selv om George Frederic Still (Hospital for sick Children, Great Ormond Street, London) allerede i 1897 beskrev forskjellene (Still GF 1897, On a form of.. reprinted in Am Dis Child 1978). Begrepene Stills sykdom, juvenil revmatoid artritt og juvenil kronisk artritt har vært brukt om barneleddgikt for å beskrive til dels overlappende pasientgrupper. “International League of Associations of Rheumatologists” (ILAR) oppnådde i 1997 enighet om å bruke samlebetegnelsen juvenil idiopatisk artritt (JIA) og definerte en rekke undergrupper. I 2019 kom forslag om ny klassifisering (Martini A, 2019). (Historisk litteratur: Schaller JG, 2005)
Etiologi og genetikk
Årsaken til barneleddgikt er i hovedsak ukjent. Basert på opphopning av familiære tilfeller og konkordans blant monozygote tvillinger på 25-40% vet vi at genetiske faktorer er av betydning (Giancane G, 2017); Hersh AO, 2015). Genetiske predisposisjoner er funnet, særlig i gener på kromosom 6 som koder for visse HLA molekyler. HLA DR8 er økt ved alle subtyper av JIA bortsett fra ved den systemiske formen. Det foreligger en 11,6 ganger økt risiko for JIA dersom et søsken har sykdommen (Prahalad S, 2010). Entesitt-relatert JIA er sterkt knyttet til HLA-B27. Hvilken rolle mikroorganismer som parvovirus B19, Epstein-Barr virus, enteritt-bakterier, klamydia pneumonia og streptokokk infeksjoner er fremdeles omdiskutert (Rigante D, 2014).
Patogenese
Gjeldende hypotese for patogenesen ved oligo- og polyartritt er at HLA molekylene som sitter på celleoverflaten, binder seg til ukjente utløsende faktorer (muligens fragmenter av virus, bakterier, andre faktorer i omgivelsene eller auto-peptider derivert fra brusk eller annet leddrelatert vev). HLA bundet antigen binder seg til T-lymfocytter og aktiverer disse. Det fører til øket produksjon av signalmolekyler, cytokiner, Th1 (IFN-gamma utsondrende T celler) og Th17 (interleukin -17 utsondrende T celler) fra det adaptive immunsystemet adaptive er av størst betydning for de fleste IJA subtypene. IL-17 induserer proinflammatoriske cytokiner og matriks metalloproteinaser, som medfører leddskade ved oligoartritt, polyartritt og psoriasis artritt (Lin Y-T, 2011). Ved entesitt relatert artritt er IL-23 et viktig cytokin som medfører inflammasjon via IL-17 og tumor nekrose faktor (TNF), mens ny benformasjon initieres av IL-22. (Kehl S, 2015). Ved systemisk JIA er derimot aktivering av det innate immunsystemet. Aktiverte monocytter, makrofager og neutrofile leukocytter forårsaker utsondring av proinflammatoriske i form av IL-1 beta, IL-6, og IL-18 (Mellins ED, 2011). Kronisk inflammasjon i leddene medfører akkumulering av synovial væske og fortykkelse av synovialt vev. Histologisk foreligger synovial hyperplasi og infiltrasjon av ulike inflammatoriske celler. Prosessen medfører at pannus dannes med påfølgende skade på brusk og benvev (McMahon A-M, 2011).
Epidemiologi

Juvenil idiopatisk artritt er den vanligste kroniske revmatiske sykdommen blant barn. En årlig insidens av barneleddgikt på 14-23 per 100.000 barn under 16 år er funnet i epidemiologiske studier fra Norge og andre skandinaviske land. Internasjonalt er det i populasjonsbaserte studier funnet gjennomsnittlig 13 nye tilfeller per 100 000 barn per år. Det betyr at det er 140-200 nye per år i Norge, og ca. 1-2/1000 (0,1-0,2%) av alle barn får barneleddgikt (Horneff G, 2022). Sykdommen kan oppstå i alle aldre, men de ulike subgruppene av sykdommen har sine typiske debut-aldersgrupper. Generelt foreligger en topp ved 1-3 års alder og en ved 9 års alder. Jenter rammes samlet sett oftere enn gutter. Foredlingen av subgrupper av JIA er: Oligoartritt (utgjør 50-80% av JIA); 2) revmatoid faktor (RF) positive polyartritt (2% – 7%); 3) RF negativ polyartritt (11% -28%) 4) systemisk artritt (4%-20%); 5) psoriasis artritt (2% -15%); 6) entesitt-relatert artritt (1% -7%) (Weiss JE, 2007).
Symptomer
Artritt-manifestasjonen ved JIA medfører oftest smerte, hevelse og stivhet med varighet mer enn 30 minutter. Symptomene er tydeligst om morgenen og bedres av fysisk aktivitet. Debutsymptomer kan være halting, særlig om morgenen. Sykdommen utvikler seg gradvis. Plutselig symptomdebut er ikke typisk. Entesitt, uveitt og feber kan også være tidlige symptomer (Garner AJ, 2021).
Debut-symptomer ved JIA kan være smerter, men barn har redusert evne til å rapportere og lokalisere smerte. Hyppigst starter sykdommen med halting, vegring mot å bruke en ekstremitet eller redusert bevegelighet i ett ledd. Hevelse, oppdrivning og feilstilling kan sees. Oftest affiseres knær og/eller ankler. Barnet kan ha allmenn-symptomer, feber, utilpasshet og irritabilitet. Febertopper over 39 grader sees ved den systemiske formen, mens lavgradig konstant feber kan sees ved polyartritt. Om lag 20% av barna utvikler en stum uveitt. Barneleddgikt kan videre medføre generelle eller lokaliserte vekstforstyrrelser med kortvoksthet, benlengde-forskjell, deformiteter eller nedsatt bentetthet.
Uveitt er en relativt vanlig komplikasjon i nordeuropeisk befolkning, sjelden blant etnisk afrikanere, latin-amerikanere og asiater (Consolaro A, 2019). Uveitt kan oppstå før, samtidig eller etter artritt-debut og trenger ikke gi åpenbare symptomer. Oligoartritt med ANA antistoff (uten subgrupper) er disponerende. Fordi symptomer ofte er være fraværende må utsatte pasienter screenes regelmessig, vanligvis hver 4-6 måned, av øyelege.
Kjeveledd: Spesielt ved oligoartikulær- og polyartikulær former for JIA angripes kjeveleddene hos mer enn 30%. De som hyppigst får kjeveledd-artritt er jenter, oligoartritt og ANA-positive (von Schuckmann L, 2020). Typiske symptomer er smerte ved tygging og ømhet ved palpasjon av leddet. Ved undersøkelse er gapeevnen (for eksempel avstanden mellom tannrekkene) redusert og underkjeven devierer mot den siden som er mest affisert. Gullstandard for undersøkelse er MR med kontrast. Systemisk behandling og i noen tilfeller injeksjon i leddet forebygger utvikling av vekstforstyrrelser med mikrognati i kjeven.
Alder ved debut, antistoff (ANA, a-CCP, RF), genetikk (HLA-B27) og kliniske manifestasjoner variere mellom de ulike undergruppene av JIA. Disse er omtalt i egne avsnitt nedenfor og sammenfattet i tabellen:
JIA subtyper (etter Stoll ML og Cron RQ, Pediatric Rheum, 2014) |
||||||
| Systemisk JIA | Oligoartikulær | RF- Polyartikulær | RF+ Polyartikulær | Entesitt relatert | Juvenil Psoriasis artritt | |
| Vanligste alder v. debut | 2-3 år | 1-3 år | To topper | Tenåringer | Tenåringer | To topper |
| Kjønn | Lik | Jenter>Gutter | Jenter>Gutter | Jenter>Gutter | Gutter>Jenter | Jenter>Gutter* |
| ANA+ | Sjelden | De fleste | De fleste | Sjelden | Sjelden | Blant de yngste |
| RF/CCP+ | Nei | Nei | Nei | Ja | Nei | Nei |
| HLA-B27+ | Nei | Nei | Nei | Nei | De fleste | De eldste |
| Uveitt | Sjelden | Ja, asymptomatisk | Ja, asymptomatisk | Sjelden | Typisk akutt | Ja, asymptomatisk |
| Entesitt | Nei | Nei | Nei | Nei | Ja | Blant de eldste |
| Daktylitt | Nei | Sjelden | Nei | Nei | Ja | Ja |
| Feber | Høye febertopper | Nei | Nei | Nei | Nei | Nei |
| *gjelder de yngste | ||||||
Utredning
Anamnesen kartlegger aktuelle symptomer (se ovenfor). Symptomdebut, alder, feber-forløp, erythem, eksantem og antall angrepne ledd er av betydning.
Klinisk undersøkes systematisk i henhold til rutiner for barn (vennligst se eget kapittel om undersøkelser hos barn og unge). Antall angrepne ledd med lokalisering noteres. Kjeveledd, hud og øyne vurderes.
Laboratorieprøver. Ved nyoppstått artritt hos barn tas ofte: CRP, SR, Hb, hvite med differensialtelling, trombocytter, ferritin, Borrelia-antistoff, AST, ANA, RF, anti-CCP, blodkultur ved høy feber eller annen mistanke om sepsis, samt urin stiks. Antistoff mot yersinia og avføring til dyrkning tas ved aktuelle tarm-symptomer. Antistreptolysin titer (AST) ved mistanke om streptokokkindusert sykdom. HLA-B27 rekvireres ved mistanke om entesitt-relatert JIA eller reaktiv artritt (som er sjelden hos små barn). Ferritin, fibrinogen og triglyserid er aktuelle ved spørsmål om makrofag aktiverings syndrom (MAS).
-Immunologiske tester. Om lag 30% har antistoffer mot egne cellekjerner (ANA), som ikke lar seg spesifisere nærmere. Positiv revmatoid faktor og/eller anti-CCP påvises hos under 10%.
Leddvæske. Ved ny monoartritt eller der det ellers er behov for å utelukke infeksjon tas leddvæske til dyrkning og PCR-undersøkelser. Mer om leddvæske-undersøkelser i eget kapittel
Bildediagnostikk. Ulike former for bildediagnostikk benyttes. Konvensjonell røntgen fremstiller skjelettet godt, men tidlige artritt-forandringer påvises ikke. Røntgenundersøkelse av affisert(e) ledd kan likevel være viktig for differensialdiagnostisk vurdering, med tanke på tidlig ledd-destruksjon (særlig ved infeksiøs artritt) og vekstforstyrrelser. Inflammatorisk artritt medfører initialt ofte en overvekst av affiserte epifyser som kan ses radiologisk. Senere lukker epifysene seg tidlig, slik at benveksten stanser for tidlig.
-Ultralyd er en rask, lite belastende og ukomplisert metode som brukes mye for å demonstrere økt væskemengde og/eller synovitt og or å veilede injeksjoner. Undersøkelsen egner seg bra hos barn og en kan evaluere alle perifere ledd. Et leddspesifikt skåringssystem med klinisk nyttig referanseatlas for juvenil artritt er tilgjengelig (Sande NK, 2021).
-Konvensjonell røntgen kan vise større leddskader ved langvarig sykdom eller vekstforstyrrelser.
-MR-undersøkelser er sensitiv for påvisning av hydrops og den mest nøyaktige metoden for vise synovitt (Dimitrou C, 2017). Benmargsødem er tidlig artritt-tegn. Usurer/erosjoner påvises tidligere enn ved konvensjonell røntgen (Kirkhus E, 2011). Differensialdiagnostisk kan MR være en viktig metode for å påvise fordeling av benmargsødem som f. eks. ved osteomyelitt eller leddnære svulster.
Biopsi. Dersom en tar vevsprøve fra synovial-membranen i et JIA-ledd, forventes histologisk høy forekomst av infiltrerende inflammatoriske celler som T- og B lymfocytter, plasmaceller, makrofager og dendrittiske celler. Leddhinnen beskrives som villøs hypertrofisk med hyperplasi av synoviocytter, endotelhyperplasi og med tegn til økt vaskularisering. Disse funnene er ikke spesifikke og gjenfinnes også ved revmatoid artritt (Maggi L, 2019).
Differensialdiagnoser
Inflammasjon/infeksjon
- Borrelia-artritt
- Myositt, oftest dermatomyositt blant barn
- Osteomyelitt
- Septisk artritt
- Serøs coxitt («transistent synovitt»)/toksisk synovitt
Non-inflammatorisk tilstander
- Epifysiolyse / Slipped Capital Femoral Epiphysis
- Femoroacetabulær inneklemming (femoroacetabular impingement)
- Hofteleddsdysplasi
- Legg-Calve-Perthes
- Leukemi
- Osteonekrose
- Severs sykdom (apofysitt i calcaneus)
- Traume
Vennligst les mer om differensialdiagnoser i kapitlet om Barn som halter
Behandling
De siste 15-20 årene har nye behandlingsmuligheter bedret sykdomsforløpet betydelig. Behandlingsmål er å oppnå remisjon/Inaktiv sykdom med eller uten medikamenter. Remisjon kan defineres som fravær av artritt, feber, utslett, serositt, splenomegali, uveitt, hovne lymfeknuter assosiert med JIA og normal CRP, SR, og legens vurdering av fraværende sykdomsaktivitet (Wallace CA, 2011). Hos omtrent 50% oppnås remisjon (Shoop-Worrall SJW, 2017). Det er en fordel å begynne behandlingen tidlig, forutsatt at andre, lignende sykdommer er utelukket.
Medikamenter.
NSAIDs. Ikke-steroide antiinflammatoriske midler (NSAIDs) som ibuprofen og naproksen brukes fortsatt, men sjeldnere enn før csDMARDs som metotreksat og biologiske legemidler kom på markedet. Paracetamol og NSAIDs lindrer symptomer, men vil ikke endre forløpet av JIA.
Kortikosteroider som prednisolon brukes vanligvis ikke lenger på grunn av bivirkninger som blant barn omfatter redusert vekst og osteoporose.
csDMARDs. Til forskjell fra tidligere, supplerer en raskt med et sykdomsmodifiserende medikament (DMARD), oftest metotreksat, dersom risiko for leddskade foreligger.
Biologiske legemidler. Som tredje linje behandling ved utilstrekkelig effekt, brukes biologiske legemidler, oftest anti-TNF midler ved artritt eller spondyloartritt og IL-1 hemmeren anakinra og canakinumab mot systemisk JIA.
Leddpunktikon/artrocentese og injeksjon med langtidsvirkende kortikosteroid (triamcinolon hexacetonide,= Lederspan) er en viktig behandling av JIA. Slik behandling fjerner synovitt, lindrer smerte og letter fysioterapi og andre tverrfaglige tiltak. Injeksjonene gjøres ofte ultralyd-veiledet og fører til remisjon av artritt i aktuelle ledd i mer enn 6 måneder hos ca. 80%.
Svangerskap
Fertiliteten er vanligvis ikke påvirket, og de fleste svangerskap forløper ukomplisert (Østensen M, 1991). Sammenlignet med friske har kvinner med juvenil revmatisk sykdom, inklusiv JIA, likevel økt forekomst av tidlig fødsel, relativt små nyfødte (små for gestasjonsalder), og preeklampsi (Remaeus K, 2017; Feldman DE 2016; Østensen M, J Rheumatol 2000. En populasjons-basert studie fra Sverige bekreftet at risikoen for for tidlig fødsel ved JIA var økt med odds ratio (OR) på 1,74 generelt. Risikoen økte til OR 4,12 dersom sykdommen var aktiv inn i voksen alder. Risiko for nyfødte som var små for alder var OR 1,84. Tidlig pre-eklampsi hadde en risiko på OR 6,28, mens tilsvarende for sen pre-eklampsi var 1,96 (Remaeus K, 2017). Disse studiene viste ikke økt forekomst av spontanaborter noe derimot en eldre britisk studie kunne tyde på (Packhard JC, 2002). Samlet sett indikerer studier og erfaring at gravide med aktiv JIA bør følges opp i forhold til økt forekomst av pre-eklampsi og tidlig fødsel. Ved JIA går ofte sykdomsaktiviteten ned under svangerskap, men øker igjen etter fødsel. Blant voksne som har hatt de sjeldne juvenile former for systemisk bindevevssykdommer og vaskulitt er det spesielt viktig at sykdommen er i en stabil fase i flere måneder før svangerskap og at det ikke er alvorlige manifestasjoner i indre organer. Generelt er det viktig med konsultasjon I god tid før svangerskap for å avklare aktuell medikasjon og øvrige risikofaktorer. Vennligst les mer om svangerskap og revmatisk sykdom i eget kapittel. Info fra NKSR.
Retningslinjer
ACR (American College og Rheumatology)
- Onel KB, 2022
- Ringold S, 2019: Non-systemisk polyartritt, sakroiliitt og entesitt)
- Ringold S, 2013: Systemisk JIA
- Angeles-Han ST, 2019: Uveitt ved JIA
- Wallace CA, 2011 (kriterier for remisjon)
Helsebiblioteket: Pediatriveileder
Norsk Revmatologisk Forening:
Prognose
JIA har varierende, til dels uforutsigbart forløp avhengig av subgruppe og individuelle forhold. Som en tommelfingerregel kan man si at omtrent 50% av tilfellene går i varig remisjon i løpet av oppveksten (Hersh AO, 2015). Forskjellig prognose kan delvis forutses ved spesielle fenotyper, genetiske disposisjoner, patogenese, laboratorieprøver og sykdomsforløpet. Noen pådrar seg alvorlig sykdom med ødeleggende konsekvenser for fysisk utvikling, fremtidig utfoldelse og aktivitetsnivå. Viktig er forebygging av vekstforstyrrelser og for sterk mental belastning, samt tidlig diagnostikk av uveitt. JIA kan fortsette med aktiv inflammasjon inn i voksen alder (Hersh AO, 2015).
Klassifikasjon
Ved juvenil oligo- og polyartritt skal leddbetennelse ha vart minst 6 ukers og alle former skal debutere før 16 års alder. Artritt defineres som hevelse i ledd eller nedsatt leddbevegelighet kombinert med ømhet / varme, som ikke kan forklares av annen sykdom eller skade. Sykdommen karakteriseres av synovitt, betennelse i leddhinnen og ekstra-artikulære manifestasjoner. ILAR (International Leage of Associations for Rheumatology) definerte i 2001 syv grupper JIA: 1) Oligoartritt; 2) revmatoid faktor (RF) positive polyartritt; 3) RF negativ polyartritt; 4) systemisk artritt; 5) psoriasis artritt; 6) entesitt-relatert artritt; 7) udifferensiert artritt (Petty RE, 2004). Siden 2019 foreligger forslag til ny klassifikasjon der “early onset ANA positiv artritt” er definert som en egen gruppe (Martini A, J Rheum 2019). Vennligst les mer om klassifiseringene nedenfor i dette kapitlet.
Klassifisering av JIA har betydning for patogenese, behandling og prognose. Ulike betegnelser er brukt. ILAR-klassifikasjoner har vært mest brukt de senere år. Det forventes at de nye (2019) forslåtte kriterier vil overta gradvis. Mer om klassifisering nedenfor i dette kapittelet.
- American Collage of Rheumatology (ACR) har klassifisert Juvenil revmatoid artritt (JRA) i tre hovedgrupper: Systemisk, Polyartikulær- og Pauciartikulær JIA
- EULAR: Juvenil kronisk artritt (JCA): Systemisk-, Polyartikulær-, Pauciartikulær- JICA. Juvenil psoriasis artritt, Juvenil ankyloserende spondylitt
- International Leag Against Rheumatism (ILAR): Juvenil Idiopatisk artritt (JIA): Systemisk-, Polyartikulær RF-, Polyartikulær RF+, Oligoartikulær persisterende, Oligoartikulær utvidet JIA. Psoriasis artritt. Entesitt relatert artritt.
- Martini A, 2019-kriterier

Systemisk Juvenil Artritt (sJIA)
ICD-10: M08.2
Definisjon: Systemisk form for JIA (sJIA) skiller seg vanligvis tydelig fra de andre gruppene barneleddgikt når det gjelder patogenese, symptomer, sykdomsforløp og behandling. Det er forslått å klassifisere sJIA blant de autoinflammatoriske sykdommene (periodiske febersyndromer). Debut hos voksne omtales som adult Stills sykdom.
Historie: sJIA ble initialt omtalt som Stills sykdom. Bakgrunnen er at den britiske pediater og patolog Georg Frederic Still i 1897 beskrev hvordan artritt hos barn skilte seg fra tilsvarende hos voksne og mange av hans beskrevne tilfeller hadde høy feber og påfallende lite destruktiv artritt, forenelig med sJIA eller dagens klassifisering. Sykdomsdebut tilsvarende sJIA hos voksne betegnes Adult Stills sykdom.
Genetikk: Genetiske variasjoner ved sJIA overlapper med tilsvarende for genetiske autoinflammatoriske sykdommer (Hinks A, 2013; Lotfy HM, 2014). Disse omfatter SNPs i interleukin-1 ligand cluster, TNFRSF1A og MVK gener.
Patogenese: Det innate immunsystemet spiller en sentral rolle i sykdomsutviklingen av sJIA, slik som ved de autoinflammatoriske sykdommene/periodiske febersyndromer. Interleukin-1 og 6 (IL-1 og IL6) bidrar betydelig til systemisk inflammasjon og er sentrale mål i systemisk behandling av SJIA.
Epidemiologi: sJIA utgjør 4-15% av tilfellene med juvenil artritt. Forekomsten er likt fordelt mellom jenter og gutter og kan debutere i alle aldre (Adult onset Stills blant (unge) voksne) (Glancane G, 2016).
Symptomer: Høye febertopper, oftest >39 grader 1-2 ganger daglig over minst tre dager, med spontan normalisering av temperatur (uten febernedsettende medikasjon) i mellom er typisk og med en varighet over minst to uker.
Artritt kan være mindre fremtredende ved debut, men ofte symmetrisk og polyartikulær senere i sykdomsforløpet.
Eksantem. Et lyst (lakse-) rødt flyktig, flekket utslett, kan ses under feberepisoder i en del av tilfellene.
Noen har sterke abdominale smerter (peritonitt forekommer), thoraks-smerter (pleuritt, perikarditt kan ses) og myalgi, delvis kombinert med feberstigning.
Kliniske kjennetegn: Feber-episoder, forstørrede, uømme lymfeknuter hos mer enn 25%, hepato-splenomegali og/eller perikarditt, pleuritt eller peritonitt kan påvises ved høy inflammasjon. Artritt. Sårt svelg er vanlig symptom hos de eldste barna.
Laboratorieprøver: Ingen spesifikk test. Høye inflammasjons-parametere (SR, CRP, trombocytose) og økt antall (neutrofile) leukocytter (20-30.000/mm3 er ikke uvanlig), høyt ferritin, men ingen auto-antistoffer i blodprøver forventes og ingen tester er spesifikke. Mikrocytær anemi er oftest mer uttalt enn ved andre kroniske inflammatoriske sykdommer.
Makrofag Aktiverings-Syndrom (MAS)
I alt utvikler ca. 10% av barna med sJIA Makrofag Aktiverings-Syndrom (MAS). Komplikasjonen ses også ved juvenil SLE og der sykdommene kompliseres av virusinfeksjon, særlig Epstein-Barr virus (EBV). Vennligst les om MAS i eget kapittel.
Differensialdiagnoser til sJIA: Ved sykdomsdebut er ingen sikker diagnose mulig. En må utelukke bakterie- eller virusinfeksjon, malignitet (leukemi) og andre revmatiske sykdommer (Kawasaki sykdom). Ved mulig eksponering må malaria vurderes. Her er en huskeliste (Tilpasset etter Giancane G et al, Rheum & Therapy 2016)
- Autoinflammatoriske sykdommer (TNFAIP3 mutasjon, PFAPA med flere)
- Castleman sykdom
- DaDA2, polyarteritis nodosa
- Inflammatorisk tarmsykdom: Crohns sykdom
- Infeksjoner: Bakterier/sepsis. mykoplasma, bakteriell endokarditt, Tyfoid feber. Malaria. Leishmaniasis. Brucellose. Virale infeksjoner
- Malign sykdom: Leukemi. Lymfom. Nevroblastom
- MIS-C: Multiorgan inflammatorisk sykdom ved SARS-CoV-2. Se også differensialdiagnoser ved makrofag aktiveringssyndrom (MAS) (Hu Y, 2021)
- Revmatisk feber
- Systemiske bindevevssykdommer: Juvenil SLE
- Systemisk vaskulitt: Kawasaki syndrom
Behandling av SJIA: Mot symptomer er det vanlig å gi NSAIDs som naproksen eller ibuprofen. Den videre behandlingen har endret seg de siste årene. Tidligere var neste steg glukokortikoider (prednisolon) og csDMARDs (metotreksat). En bedre strategi har vist seg å være biologisk behandling i form av IL-1 hemmeren anakinra, alternativt canakinumab (dyrere). Et annet alternativ er IL-6 hemmer (tocilizumab). En forventer nesten umiddelbar effekt. Ved mistanke om MAS (se ovenfor) suppleres med glukokortikoider i høye doser og andre anbefalte tiltak.
Sykdomsforløp: Monosyklisk eller persisterende, sykdom ses hos over 50%. Artritt følger da ofte feberepisodene og prognosen på lengre sik er ofte god. Et annet forløp foreligger når de systemiske symptomene med feber går tilbake, men artritten blir kronisk. Denne formen kan medføre betydelige destruksjoner i ledd.
Behandling med IL-6 hemmer (tocilizumab) eller IL-hemmer (anakinra, canakinumab) har i enkelte tilfeller medført overømfintlighets-reaksjon (hypersensibilitet) med nytt utslett, lunge-manifestasjon, eosinofili, lymfopeni og høy ferritin i blodet (Saper VJ, 2019).
Klassifikasjonskriterier: Vennligst se nederst i dette kapittelet.

Oligoartikulær / pauciartikulær JIA
ICD-10; M08.4
Definisjon: Sykdommen omfatter fire eller færre (<5) ledd i løpet av sykdommens seks første måneder. ILAR definerer to undergrupper: Ekstendert oligoartikulær JIA dersom flere ledd angripes etter seks måneders forløp og persisterende oligoartikulær JIA dersom forløpet begrenses til fire eller færre (<5) ledd. Oligoartikulær/pauciartikulær JIA (etter ILAR-klassifikasjonen) tilsvarer i stor grad med “Early-onset ANA-positiv JIA” etter Martini 2019-klassifikasjon (se nedenfor). Denne formen for artritt debuterer ikke hos voksne.
Genetikk: RF / CCP negativ oligoartikulær JIA er assosiert med HALDRB1:11:03/04 og DRB1:08:01. En beskyttende effekt er beskrevet ved genet DRB1:15:01 (Hersh AO, 2015).
Epidemiologi: Oligoartikulær JIA utgjør 50-80% av alle med JIA. De fleste er jenter. Sykdommen kan debutere i alle barne-årene, men hyppigst ved 2-3 års alder og sjelden etter 10 års alder. Omtrent 50% utvikler ekstendert sykdom (se ovenfor).
Symptomer på Oligoartikulær artritt
Ved sykdomsdebut observeres at barnet halter om morgenen. Smerter er mindre fremtredende, selv om et kne, ankel eller annet ledd kan være hovent og noe varmt. Artritt i ett kne (30-50%) eller en en ankel er vanligst. Asymmetrisk affeksjon er vanligst. Artritt i håndledd og ankler kombinert med høy senkningsreaksjon (SR) og CRP indikerer økt risiko for ekstendert sykdom (Al-Matar MJ, Petty RE, 2002). Hofteledd angripes sjelden (differensialdiagnose: infeksjon) og feber er uvanlig.
Klinisk undersøkelse ved Oligoartikulær artritt
Artritt-tegn i ett eller flere ledd (kjeveledd/gapeevne og cervical-columna bør også vurderes). Redusert bevegelighet i ledd kan påvises. Vekstforstyrrelser som benlengdeforskjell, skoliose og mikrognati vurderes. Kontroller hos øyelege er viktig.
Nærmere om anamnese og undersøkelser er beskrevet i eget kapittel
Laboratorieprøver: CRP, SR og trombocytter kan være moderat forhøyet. ANA foreligger hos ca. 80% og er assosiert med risiko for uveitt. Spesifikke ANA-undergrupper forventes ikke.
Bildediagnostikk: Ultralydundersøkelser er godt egnet for diagnostikk og kontroll i tillegg til klinisk vurdering. MR dersom behov for nærmere avklaring. Røntgen ved vurdering av vekstforstyrrelser.
Differensialdiagnoser ved Oligoartikulær artritt
- Artrogryposis multiplec congenita (en gruppe medfødt genetiske tilstander med ledd kontraktur i to eller flere kroppsregioner (Dahan-Oliel N, 2019)
- Barnemishandling ved mulige traumatiske leddskader
- Borreliose
- Hemofili (blødning, hemartrosis)
- Idrettsskade
- Infeksjoner, andre: tuberkulose, annen septisk artritt, osteomyelitt
- Juvenil artritt, andre typer
- Leukemi (ALL) eller annen malign sykdom (nevroblastom, lymfom)
- Osteoid osteom
- Osteonekrose, steroid-indusert
- Pigmentert villonodulær synovitt
- Reaktiv artritt etter streptokokk-infeksjon
- Revmatisk feber
- Sigdcelle anemi
- Skjørbuk (C-vitamin mangel)
- Toksisk synovitt
- Tumor i skjelett (leddnært: osteosarkom)
Sykdomsaktivitet og behandlingsmål defineres før behandlingsstart. Barnet og foresatte informeres også om medikamentene, for eksempel via pasientinformasjonsark.
Leddpunksjon / artrocentese og injeksjon av kortikosteroid (Lederspan) har god effekt. NSAIDs forventes å ha supplerende effekt. Ved god behandlingseffekt kan begrenses til disse.
csDMARDs som metotreksat eller andre sykdomsmodifiserende legemidler gis ofte dersom fem eller flere ledd angripes eller sykdommen ikke er tilstrekkelig behandlet med DMARDs og artrocentese. Biologiske legemidler brukes sjelden ved oligoartikulær sykdom, men kan suppleres ved et aggressivt forløp eller når uveitt foreligger (etanercept har ikke effekt på uveitt, adalimumab foretrekkes).
Prednisolon og andre systemiske kortikosteroider er ikke en etablert del av behandlingen.
Biologiske legemidler benyttes i alvorlige tilfeller, oftest der mange ledd er angrepet i forløpet (ekstendert type).
Prognose: Over 50% av tilfellene er selvbegrensende innen 5 år (Guzman J, 2015). Ekstendert sykdom (se ovenfor) har generelt dårligere prognose enn persistent type. Veksthemming forekommer. Blant barn med øye-komplikasjon / uveitt kan varig redusert syn forekomme hos ca. 15% (Consolaro A, 2015).
Klassifikasjonskriterier: Vennligst se nederst i dette kapittelet.
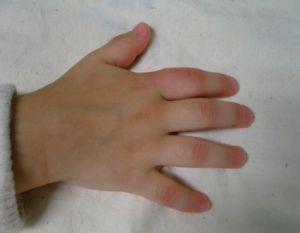
Polyartikulær JIA
ICD-10: M08.3 (seronegativ)
Definisjon: Polyartikulær JIA angriper fem eller flere ledd de første 6 måneder av sykdomsforløpet. Sykdommen deles inn i to undergrupper: Seronegativ og seropositiv polyartikulær JIA, uten eller med RF og anti-CCP antistoff. Den seropositive typen ligner mest på revmatoid artritt (RA) hos voksne.
Seronegativ polyartikulær JIA
Denne formen omfatter ca. 25% av JIA og debuterer oftest ved 2-5 års alder. Revmafaktorer (RF) og CCP antistoff er fraværende, men ANA (uten subgrupper) påvises hos mange. Sykdommen har klare fellestrekk med oligoartikulær artritt også ved at jenter angripes hyppigst, og de har en høy forekomsten av uveitt/iridocyklitt. Artritt er ofte symmetrisk og omfatter ikke sjelden fingre og tær. GWAS (genetikk/genom-studier) har ikke påvist forskjeller i sykdomsdisposisjon mellom seronegativ polyartikulær JIA og oligoartikulær JIA, noe som styrker hypotesen om at de er to utgaver av samme sykdom. Øyelegekontroller anbefales som ved oligoartikulær form.

Seropositiv polyartikulær JIA
Denne formen debuterer oftest etter ti års alder og er hyppigst hos jenter. En påviser RF og CCP-antistoff i blodprøver og sykdommen er ganske lik revmatoid artritt hos voksne. Små ledd i fingre og tær, håndledd, albuer, skuldre, nakke, kjeveledd, hofter, knær og ankler angripes, ofte samtidig på begge sider (symmetrisk artritt). Uveitt/iridocyklitt ses sjelden, men regelmessig øyelege-kontroll (hver 6. måned) anbefales likevel. Sykdommen og forløpet er som ved revmatoid artritt hos voksne. En må dermed regne med behov for medikamentell behandling i mange år.
Differensialdiagnoser:
- Barnemishandling dersom tegn på traumatisk leddskade
- Borrelia
- CRMO (non-bakteriell multifokal osteomyelitt)
- Farber lipogranulomatose (metabolsk lagringssykdom)
- Hypermobilitetssyndrom
- Inflammatorisk tarmsykdom med artritt
- Revmatisk feber
- Sarkoidose/Blau syndrom
- Skjørbuk (C-vitamin mangel)
- Smertesyndromer
- Systemiske bindevevssykdommer (JSLE, Sjøgrens)
Behandlingen er både for seronegativ og seropositiv type med NSAIDs, kortison (Lederspan) -injeksjoner i affiserte ledd, DMARDs (metotreksat) og ofte suppleres med biologiske legemidler.
Prognosen er vanskelig å forutsi. Høy sykdomsaktivitet de første to år fra debut, hyppige feberepisoder, mange angrepne ledd og anti-CCP antistoff regnes som risikofaktorer for alvorlig forløp (Prieur A-M, 2001). I tillegg er naturlig nok, behandlingsresponsen avgjørende.
Klassifikasjonskriterier: Vennligst se nederst i dette kapittelet.
Entesitt-Relatert Artritt (eRA)
ICD-10: M08.1
Definisjon: Entesitt-relatert artritt tilsvarer juvenil ankyloserende spondylitt (Bekhterevs) affiserer enteser (senefester) og iliosakralledd. De fleste har vevstypen HLA-B27. Tilstanden overlapper og er delvis identisk med spondyloartritt hos barn. Illustrasjon: Burgos-Vargas R, 2012. CC BY 2.0.
Epidemiologi: eRA utgjør 10-20% av JIA og 60-80% er gutter, de fleste er over 12 år alder ved debut, men tilstanden kan ses fra seks års alder.
Patogenese og genetikk: Det foreligger en sterk assosiasjon med HLA-B27. Det er mange ganger økt risiko for eRA dersom Bekhterevs / ankyloserende spondylitt foreligger blant første grads slekt (foreldre, søsken). Samtidig forekomst av Ulcerøs kolitt, Crohns sykdom eller psoriasis ses hos noen.
Symptomer på eRA
Artritt omfatter vanligvis knær, ankler og hofter. Omtrent halvparten får artritt i fire eller færre ledd. Daktylitt i fingre og tær. Entesitt foreligger oftest ved festet for Akillessenene på calcaneus, patellarsener ved knær og plantarfascier. Halting ved entesitt eller artritt er vanlig. Det er viktig også å diagnostisere uveitt / iridocyklitt som kan være asymptomatisk. Øyelege-screening anbefales derfor. Inflammatorisk ryggsmerte kan indikere begynnende manifestasjoner i iliosakralledd og columna. Iliosakralledd (IS) angripes i forløpet, men symptomer fra IS-ledd er vanskelige å verifisere før langt opp i tenårene. Columna angripes (syndesmofytter, ankylose) senere i forløpet og ses sjelden i barnealder.
Bildediagnostikk: Inflammasjon i i enteser og IS ledd visualiseres tidligst med ultralyd (enteser) og MR (enteser og IS-ledd). Fordi MR ikke skiller godt mellom normale vekstprosesser og artritt i IS ledd kan tidlige forandringer i IS-leddinflammasjon lett feiltolkes/overdiagnostiseres. Patologiske forandringer ses først på iliumsiden av IS-leddene. Typisk er ødem i leddnære skjelettområder med utvikling av usurer i leddflatene. Inflammatoriske forandringer i columna og ankylose i IS-ledd og columna kan utvikles senere i sykdomsforløpet. Vennligst les mer om bildediagnostikk i eget kapittel
Differensialdiagnoser:
- Barnemishandling ved tegn på traumer
- CRMO
- Inflammatorisk tarmsykdom (IBD) med artritt
- Halting av andre årsaker
- Overbelastning av patellarsener
- Schlatters sykdom (apofysitt og osteonekrose)
Behandling
- NSAIDs som ibuprofen eller naproksen brukes som symptomlindrende tiltak.
- Ved perifer artritt er leddpunksjon og kortikosteroid-injeksjon (Lederspan) aktuelt.
- Ved overveiende perifer artritt vurderes DMARDs (metotreksat), men effekt på det aksiale skjelett (inklusiv IS-ledd) forventes ikke.
- Biologiske legemidler som adalimumab, etanercept eller infliksimab forventes å ha god effekt også på columna og IS-ledd når det foreligger aktiv inflammasjon.
- Fysioterapi for å bedre og opprettholde bevegelse, styrke og kroppsholdning og annen tverrfaglig behandling er viktig.
Klassifikasjonskriterier: Vennligst se nederst i dette kapittelet.
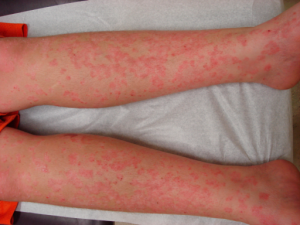
Juvenil Psoriasisartritt (JPsA)
ICD-10: M09.0; L40.5 (psoriasis med artropati)
Psoriasisartritt hos barn er en vanskelig diagnose som kan overlappe med andre former for JIA. Relativt spesifikke symptomer er psoriasiseksantem, negle-pitting og daktylitt.
Noen har entesopati, men da en uklar avgrensing mot entesitt-relatert JIA. Artritten kan være asymmetrisk og ANA relatert, noe som er felles med oligoartikulær JIA. Der RF og CCP-antistoff forekommer er sykdommen forenelig med seropositiv polyartritt. Psoriasisartritt hos voksne er beskrevet i eget kapittel.
Klassifikasjonskriterier: Vennligst se nederst i dette kapittelet.
Udifferensiert juvenil artritt
ICD-10: M08.9
I tidlig sykdomsfase kan differensiering være vanskelig hos mange, men i forløpet klassifiseres de fleste. Når JIA foreligger, men tilstanden passer flere av subgruppene på grunn overlappende symptomer og undersøkelsesfunn, kan sykdommen klassifiseres som udifferensiert JIA. Dette tilsvarer de udifferensierte former for revmatisk sykdommer blant voksne. Håndtering av slike tilfeller tilpasses individuelt. Man retter behandling og oppfølging etter den JIA-subgruppen som ligger nærmest de viktigste symptomer og undersøkelsesfunn. Klassifikasjonskriterier: Vennligst se nederst i dette kapittelet.
ILAR-klassifisering av JIA
Systemisk JIA
Artritt i minst ett ledd og forutgående feber dokumenterbart i minst tre dager over minst to ukers varighet. I tillegg skal minst ett av følgende være assosiert:
- Flyktig utslett
- Generalisert lymfadenopati
- Hepato/splenomegali
- Serositt
- Eksklusjon ABCD: se nedenfor
Oligoartikulær
Persisterende eller ekstendert oligoartritt:
- Artritt i 1-4 ledd de første 6 måneder av sykdomsforløpet
- Persisterende: Inntil 4 affiserte ledd gjennom hele sykdomsforløpet
- Ekstendert: Mer enn 4 ledd etter 6 måneders forløp
- Eksklusjon: ABCDE (se nedenfor)
Polyartikulære former
-RF-negativ polyartritt
- RF-
- Artritt i 5 eller flere ledd i løpet av de første 6 måneders sykdomsforløp
- Eksklusjon ABCDE (se nedenfor)
-RF positiv polyartritt
- RF+
- Artritt i 5 eller flere ledd i løpet av sykdommens 6 første måneder
- Eksklusjon ABCE (se nedenfor)
Juvenil Psoriasis-artritt
Artritt + psoriasis eller artritt og minst to av følgende assosierte funn:
- Daktylitt
- Negle-pitting
- Onycholyse
- og/eller psoriasis hos første-grads slektning (foreldre, søsken)
- Eksklusjon BCDE (se nedenfor)
Entesitt-relatert artritt
Artritt og/eller entesitt med minst to av følgende
- Påvist eller anamnese på smerter i iliosakralledd med- eller uten inflammatorisk ryggsmerte
- HLA-B27+
- Sykdomsdebut med artritt hos en over 6 år gammel gutt
- Akutt (symptomatisk) anterior uveitt
- Sykehistorie blant første grads slekt (foreldre, søsken) på følgende:
- Bekhterevs/ankyloserende spondylitt
- Entesitt-relatert artritt
- Sakroiliitt med inflammatorisk tarmsykdom (ulcerøs kolitt, Crohns)
- Reiters syndrom
- Akutt uveitt
- Eksklusjon: ADE (se nedenfor)
Udifferensiert JIA
Artritt som ikke fyller noen av de ovenfor nevnte kriterier eller som samtidig fyller to eller flere av kriteriene
Eksklusjoner (gjelder alle ILAR-kriteriene ovenfor)
A. Psoriasis eller sykehistorie på psoriasis hos pasienter eller deres første grads slektninger.
B. Artritt blant HLA-B27 positive gutter med debut etter 6 års alder
C. Bekhterevs / Ankyloserende spondylitt, entesitt-relatert artritt, sakroiliitt med IBD, Reiters syndrom, akutt anterior uveitt eller en sykehistorie på disse blant første gards slektninger
D. Forekomst av IgM revmatoid faktor (RF) ved minst to anledninger med minst tre måneders avstand E. Påvist systemisk JIA
Foreslått ny JIA- Klassifisering av 2019
A. Systemisk JIA
Feber av ukjent årsak (utelukke infeksjoner, neoplasmer, autoimmune eller autoinflammatoriske sykdommer). Feberen skal dokumentert opptre minst en gang daglig ≥ 39°C og falle til ≤ 37 °C mellom toppene og feberen skal opptre på minst tre påfølgende dager i løpet av en to ukers periode.
I tillegg kreves enten to major kriterier eller ett major og to minor-kriterier.
Major kriterier
(1) Flyktig (nonfiksert) erythematøst utslett
(2) Artritt
Minor kriterier
- Generalisert forstørrede lymfeknuter og/eller hepatomegali og/eller splenomegali
- Serositt
- Artralgi (ikke artritt) over minst to uker
- Leukocytose (≥ 15,000/mm3) med neutrofili
B. RF (Revmafaktorer) -positive JIA.
Denne formen ligner mest på revmatoid artritt (RA) hos voksne. De fleste barn som angripes er jenter som nærmer seg voksen alder.
Foreslåtte klassifikasjonskriterier (Martini A, J Rheum 2019):
- Artritt i ≥ 6 uker og
- Assosiert med to positive tester for RF som tas med minst tre måneders mellomrom eller minst en positiv antistoff-test for cyklisk citrullinert peptid (CCP)
C. Entesitt/spondylitt (SpA) -relatert JIA.
Artritten kan også være assosiert med økt risiko for rygg-affeksjon (sakroiliitt), betennelse i senefester (entesitt) og vevstypen HLA-B27 (entesitt relatert JIA). Hos noen er sykdommen assosiert med psoriasis eller inflammatorisk tarmsykdom.
Foreslåtte klassifikasjonskriterier (Martini A, J Rheum 2019):
- Perifer artritt ≥ 6 uker og entesitt eller
- Artritt ≥ 6 uker eller entesitt pluss ≥ 3 måneder med inflammatorisk ryggsmerter og sakroiliitt ved bildediagnostikk eller
- Artritt ≥ 6 uker eller entesitt pluss minst to av følgende
- 1) Smerter i iliosakralledd
- 2) Inflammatorisk ryggsmerte (jfr. definisjon av Sieper J et al, 2009)
- 3) HLA-B27
- 4) Akutt (symptomatisk) anterior uveitt
- 5) SpA blant førstegradsslektning (foreldre, søsken, barn)
D. Early-onset ANA-positiv JIA.
- Artritt ≥ 6 uker og
- Early-onset (≤ 6 år), og
- To positive ANA tester med titer ≥ 1/160 (ved lmmunfluorescens) tatt med minst tre måneders avstand.
- Eksklusjon av systemisk JIA, RF-positiv JIA og entesitt/SpA relatert JIA
E. Annen JIA
- Artritt i ≥ 6 uker
- Fyller ikke kriteriene A-D ovenfor
F. Uklassifisert JIA
- Artritt ≥ 6 uker
- Oppfyller flere av av klassifikasjonene A-D
