VASKULITT (REV 034-052)
86 Eosinofil granulomatøs polyangiitt (EGPA), Churg-Strauss vaskulitt (REV 034)
Ragnar Gunnarsson
Kjennetegn på EGPA
Terapiresistent asthma, nesetetthet, nedsatt allmenntilstand. Lungeinfiltrater og fortykkete luftvier og bihuleforandringer. Eosinofili i blod og vevsprøver. Ca. 70% er ANCA negative og resisterende vanligvis MPO-ANCA positive.
Diagnosekoder ICD-10: M30.1 Eosinofil granulomatose med polyangiitt (EGPA). Polyarteritt med lungeaffeksjon, i den tiende versjon av den internasjonale statistiske klassifikasjonen av sykdommer og beslektede helseproblemer i regi av Verdens helseorganisasjon (WHO ICD10).
Prosedyrekoder: Intravenøs infusjon: WBGM00. Infusjon med gammaglobulin: RPGM05. Intravenøs infusjon med cytostatika: WBOC05. EKG: FPFE15
ATC koder (for legemiddelstatistikk): Prednisolon: H02A B06 Immunsuppressive legemidler: L04A A. Behandling med rituksimab: L01XC02, cyklofosfamid: L01AA01, mepolzumab R03DX09, benralizumab R03DX10.
Definisjon
I 2012 på den internasjonal vaskulitt-konsensuskonferansen i Chapel Hill i delstaten North Caroline («Chapel Hill Consensus Conferance» CHCC 2012), ble definisjon av sykdomsnavnet Churg Strauss syndrome formelt erstattet med eosinofil granulomatose med polyangiitt (EGPA) (1).
EGPA er definert som småkarsvaskulitt og klassifisert med ANCA vaskulitter (AAV) som granulomatøs polyangiit (GPA) og mikroskopisk polyangiit (MPA). Dette til tross for at kun 30-40% av EGPA pasientene er ANCA positive. EGPA står i særstilling blant AAV og der sykdommens patogenese overlapper med hypereosinofile syndrom. Det er to hovedformer av EGPA som avhengig av ANCA status. Det viser seg at anti-myeloperoksidase (MPO) ANCA positive EGPA pasienter har fenotype som ligner mer på vaskulitt som er både genetisk og klinisk mer forenlig med mikroskopisk polyangiit (MPA), med sykdomsmanifestasjoner som perifer nevropati, palpable purpura, nyre-affeksjon, men noe sjeldnere lunge– og hjerteaffeksjon en ANCA negative EGPA fenotypen. Som ser ut til å være mer relatert til hypereosinofil syndrom (HES) med hovedsymptomene; astma, øvre luftveissymptomer (2).
Historikk
Churg Strauss syndrom (CSS) ble i 1951 beskrevet av Lotte Strauss og Jacob Churg under navnene «allergic granulomatosis», «allergic angiitis» og «microskopic periarteritis» (3). Fra 2012 har CSS fått navnet eosinofil granulomatose med polyangiit (EGPA) som nå anvendes i internasjonal medisinsk litteratur.
Genetikk
I en nylig publisert internasjonal genom-assosiasjonsstudie (genome-wide association study – GWAS) med 676 pasienter med EGPA og 6809 kontroller (2). Den viste sterk assosiasjon til HLA typer -DRB1*08:01, *01:03 og HLA-DQA1*02:01 kun hos MPO-ANCA positive EGPA pasienter likt og hos pasienter med MPA (2). Avvik i GPA33 genet, rs9290877, og rs11745587:A som er assosiert til C5orf56-IRF1-IL5 var kun forbundet til MPO-ANCA negative EGPA pasienter. Det var flere gener som var funnet uavhengig av ANCA status, bl.a. ved gen som koder for BIM, et Bcl2-relatert protein, som nødvendig for å styre rekke funksjoner inklusiv; programmert celledød (apoptose), homeostase i immunreguleringen, overlevelse av mastceller og avvik er forbundet til økt risiko for autoimmune sykdommer (2). Avvik i BCL211 og MIR4435-2HG (MORRBID) genene er assosierte til EGPA med høyere eosinofilcelletall og astma (2). Avvik i BIM er bl.a. forbundet til hypereosinofil syndrom (HES) (4). Gen som koder for TSLP som utløses ved betennelsestilstander fra stroma- og epitelceller og stimulerer til TH2 respons og økt andel eosinofili. Allelet assosiert til rs1837253:C hos europeiske EGPA pasienter er knyttet til astma, nesepolypper og allergisk rhinitt og økt eosinofil tall. I tillegg er rs6544802 som er assosiert til BACH2, CDK6, og GATA3 assosiert til EGPA (2).
Figur 1. ANCA status og genetikk ved eosinofil granulomatose med polyangiit. (Adoptert fra Paul A Lyons og kolleger (2)).
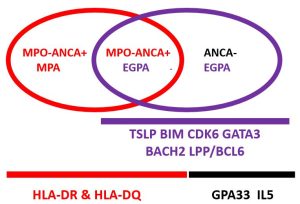
MPO : myeloperoksidase, ANCA : antinøytrofilt cytoplasma antistoff, EGPA : eosinofil granulomatose med polyangiit, MPA : mikroskopisk polyangiitt
Patogenese
Det er store vansker å avklare patogenesen for EPGA og årsakene ligger dels i at det foreligger ingen gode dyremodeller og i at tilstanden svært sjelden. I motsetning til de andre AAV som GPA og MPA, virker ikke som ANCA antistoffer har direkte rolle i patogenesen av EGPA. Som ved astma, aktiveres CD4+ T-lymfocytter hovedsakelig via Th2-type respons og med klonal ekspansjon og med frigjøring av cytokinene; IL-4, IL-5 og IL-13. Disse påvises både i EGPA-pasientenes sera og bronko-alveolær skyllevæske (BAL). IL-5 har viktig rolle i eosinofilenes modning og overlevelse, og det er en sterk assosiasjon mellom IL-5-ekspresjon og EGPA-aktivitet (BVAS og eosinofili). EGPA-patogenesen ser også ut til å implisere også Th1- og Th17-responser (5).
Klassifikasjon og sykdomsaktivitet
ANCA assosierte vaskulitter (AAV) har siste årene vært segregert til EGPA, GPA, MPA og polyarteritis nodosa (PAN) og uklassifisert vaskulitt ved hjelp av flytskjema algoritme anbefalt bl.a. av European Medicines Agency (EMA) (6). De nye og oppdaterte klassifikasjonskriterie har kommet og har avskaffet denne metoden i kliniske studier.
Figur 2. Foreslått algoritme ved klassifikasjon av ANCA-assosiert vaskulitt og polyarteritis nodosa (6).
(6). ACR: American College of Rheumatology; CHCC-2012 : Chapel Hill Consensus Conference 2012; cPAN : classic polyarteritis nodosa; EGPA : eosinofil granulomatose med polyangiitt; GPA : granulomatose med polyangiitt; MPA : mikroskopisk polyangiitt. MPO : myeloperoksiadase; PR3 : proteinase 3.
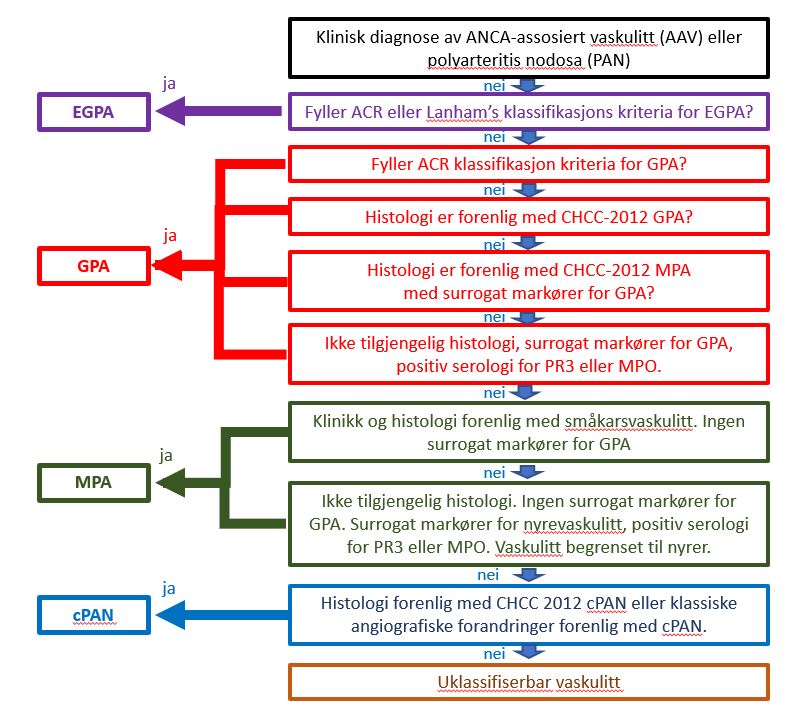
Klassifikasjon. Det er to klassifikasjonskriterier som tidligere ble anvendt for EPGA.
(1) Lanham’s kriteria fra 1984 som inkluderer at tre kriteria er oppfylt: (i) astma, (ii) perifer eosinofil (>1500/mm3 eller >10% av leukocytt tal) og (iii) påvist vaskulitt i to eller flere organer (7).
(2) American College of Rheumatology (ACR) klassifikasjonskriterier fra 1990 (8). Der må minst fire av seks av følgende kriterier være oppfylt; (i) astma, (ii) eosinofili (>10%), (iii) mono-/polynevropati, (iv) lungeinfiltrater, (v) paranasal sinus affeksjon, eller (vi) ekstravaskulære eosinofile leukocytter.
Det foreligger oppdatert 2022 ACR/EULAR klassifikasjonskriteria for EGPA. Den baserer seg på «Diagnostic and Classification Criteria in Vasculitis Study» (DCVAS) som er en internasjonal, multisenter, observasjonsstudie som har samlet data på over 1000 AAV-pasienter samt data fra pasienter med andre former for vaskulitt og pasienter med sykdommer som etterligner vaskulitt.
Figur 3. 2022 ACR / EULAR klassifikasjonskriteria for eosinofil granulomatose med polyangiitt (EGPA) (28).

BVAS. Sykdomsaktivitet ved EGPA som ved MPA og GPA vurderes etter Birmingham Vasculitis Activity Score versjon 3. Der er maksimal score på 63 poeng. Ved vedvarende sykdom er det maks. score på 33 poeng. Komplett remisjon angis som BVAS =0 med per oral Prednisolon 7,5 mg eller lavere mens vedvarende «sustained» remisjon som BVAS = 0 i 6 måneder eller mer. Kalkulator for BVAS v3 for Iphone og Android telefoner og på PDF-fil. (9, 10).
VDI. Skade ved vaskulitt vurderes etter Vasculitis Damage Index (VDI) (11). Skåring i VDI er permanent og poeng beholdes, men skaden må ha stått i minimum 3 måneder. Kalkulator VDI.
Five Factor Score. For å vurdere behandlingsstrategi må man ta stilling til alvorlighetsgrad. Den franske Five Factor Score (FSS) har vært vurdert ved ANCA assosierte vaskulitter og polyarteritis nodosa (PAN). (Tabell 1) (12, 13). Alvorlig systemisk form av EGPA har som oftest «Five Factor Score» (FFS) > 0, mens alder ≥ 65 år, ikke er alene tilstrekkelig som den eneste av de fem delene i FFS for å klassifisere EPGA som alvorlig. Annen alvorlig organaffeksjon kan også forekomme til tross for FFS=0, som lungeaffeksjon med og uten lungeblødninger og/eller nerveaffeksjon inklusiv mononevritis multipleks. Fleste bruker 1996 versjonen ved EGPA.
Tabell 1. Five-factor score for eosinofil granulomatose med polyangiitt (EGPA). (12, 13)
| Revidert 2011 «five-factor score» (12) | 1996 «five-factor score» (13) |
| Alder > 65 år | Sentralnervesystem affeksjon |
| Manglende øre-nese hals affeksjon | Proteinuri (>1 g/dag) |
| Hjerteaffeksjon med hjertesvikt | Hjerteaffeksjon |
| Nyreaffeksjon med nyresvikt (serum kreatinin > 150 µmol/L (1,7mg/dL)) |
Nyreaffeksjon med nyresvikt (serum kreatinin > 140 µmmol/L (1,6mg/dL)) |
| Affeksjon av fordøyelsessystemet | Affeksjon av fordøyelsessystemet |
Kliniske symptomer ved EGPA
A: Allmennsymptomer
")
Feber, asteni og vekttap forekommer ofte ved diagnose: hadde rundt 40% feber og myalgi, rundt 30% hadde artralgi, og nesten 50% hadde signifikant vekttap (14).
B: Lunge og øre-nese-hals (ØNH) manifestasjoner
Astma er et av kardinal symptomene og rammer flest alle pasientene (95-100%), som oftest før de utvikler tegn på systemisk vaskulitt. Det kan være sen debuterende astma, men kan også starte i barns-/ungdomsårene. Astma ved EGPA er generelt alvorlig med oftest økte symptomer ved utvikling av vaskulitt som krever økt glukokortikosteroid behandling.
Lungeinfiltrater er vanlige ved EGPA (40%), og de kan være uni- eller bilaterale, og ved oppstart av kortikosteroider vil de vanligvis forsvinne i løpet av noen dager. Alveolær blødning, forårsaket av lungevaskulitt, er sjelden ved EGPA (<5%), og kan da medføre massiv hemoptyse med uttalt dyspne, anemi og lungesvikt.
C: Nevrologiske manifestasjoner
Perifere nevropati opptrer hyppig (45-75%), der hovedpresentasjonen er mononeuritt-multipleks som skyldes vaskulitt i vasa nervorum av nerver. Pasienter kan utvikle parestesi eller smertefull hyperestesi. Kranial nerve affeksjon ser ut til å forekomme relativt sjelden (3%).
Affeksjon av hud er vanlig ved småkarsvaskulitt (40-70%). Patogenesen er enten vaskulitt i små-kar eller utvikling av ekstravaskulære granulomer med vaskulær purpura. Subkutane knuter er ofte bilaterale, symmetriske, røde. Dessverre er som regel biopsier av hudlesjoner ved EGPA ikke diagnostiske verken for EGPA eller for andre former av AAV. Andre hudmanifestasjoner kan forekomme med EGPA, deriblant urticaria, livedo reticularis og nekrotiske sår.
F: Affeksjon av gastrointestinal kanalen
Gastrointestinal (GI) involvering av EGPA er alvorlig og kommer med i five-factor score (FFS). Kliniske manifestasjoner av GI er ofte uspesifikke, fleste får abdominalsmerter, kvalme, oppkast, diaré og/eller tarmblødning. Tarmperforering, den alvorligste komplikasjonen og er forbundet med høy dødelighet.
Affeksjon av hjerte er den alvorligste EGPA-manifestasjonen og er den hyppigste årsaken til død. Hjerteaffeksjon kommer også med i five-factor score (FFS). Forekomsten av hjerte manifestasjoner ved EGPA kan lett undervurderes. I en stort fransk kohort serie som inkluderte 383 pasienter fikk 16%% og 15% av pasientene kardiomyopati og perikarditt. Det var 2,3 x større andel av de EGPA pasientene som var ANCA negative (19,2%) i forhold til de som var ANCA positive (8,3%) (14).
Pasienter med EGPA bør vurderes med ekkokardiografi og EKG og risikopasienter på scope MRI av hjerte. Og i selekterte tilfeller koronarangiografi.
H: Affeksjon av nyrer
Glomerulonefritt ved EGPA er assosiert med anti-MPO ANCA positivitet og alvorlig nyreaffeksjon er relativt uvanlig sammenlignet med andre AAV (GPA og MPA). Ved nyreaffeksjon viser nyrebiopsier pauciimmun halvmåne glomerulonefritt som ved de andre AAV.
Differensial diagnoser ved EGPA
Det er flere differensial diagnoser ved EGPA og det er multiple årsaker for eosinofili. Det kan være allergisk reaksjon av medikamenter eller tilsetningsstoffer som ved DRESS syndrom «drug reaction with eosinophilia and systemic symptoms». En hyppig årsak er infeksjoner som kan være fugal, orme, parasitt, eller viral infeksjoner. Eosinofili kan sees ved autoimmun sykdommer som; sarkoidose, IgG4-relatert sykdom og ved inflammatorisk tarmsykdom. Underliggende malignitet både solid tumorer, akutt og kronisk eosinofil leukemi og andre myeloid neoplasmer som kronisk myelogen leukemi og systemisk mastocytose. Man kan også ha primært hypereosinofil syndrom.
I våres vestlige verden er eosinofil astma, andre er parasitt infeksjoner og allergisk bronkopulmonar aspergillose. Andre tilstander må utelukkes og serologisk undersøkelse for toksokariase og utføre HIV «human immunodeficiency virus» test. Det er indisert for å teste for Aspergillus i sputum og/eller ved bronko-alveolær lavage (BAL) i tillegg bør det tas prøver for tryptase (m.h.t. mastocytose) og vitamin B12 og ta blodutstryk for å vurdere dysplasi, eosinofili eller blaster (15). Hypereosinofilt syndrom (HES), som også kan være assosiert med kardiopati, lungemanifestasjoner og nevropati. De viktigste forskjellene mellom HES og EGPA to er symptomer på vaskulitt og/eller biopsibekreftelse av EGPA. Ved HES kan man se forhøye tryptase verdier og /eller vitamin B12 nivå. Genetisk testing for FIP1L1-PDGFRA-fusjon, c-kit og JAK2-mutasjoner (16-18). Gleich syndrom som også kalles episodisk angiødem med eosinofili og systemisk mastocytose med eosinofili er også sjeldne differensial diagnoser.
Behandling
Behandlingsretningslinjer som tar for seg behandling av EGPA:
- EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update (15).
- 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis (19).
- Behandlingsanbefalinger for ANCA assosierte vaskulitter fra Norsk revmatologisk forening (NRF) fra 2024.
Eosinofil granulomatose med polyangiit (EPGA) er sjelden sykdomstilstand og det foreligger foreløpig svært begrenset med randomiserte medikamentstudier. Pasienter med ANCA positivitet (ca. 30-40%) har som oftest pANCA/MPO-ANCA. ANCA positive pasienter har oftere vaskulittiske trekk som heller ofte mer imot mikroskopisk polyangiitt (MPA).
Figur 3 – Induksjon og vedlikeholdsbehandling av eosinofil granulomatose med polyangiitt i følge Norsk revmatologisk forening (NRF).
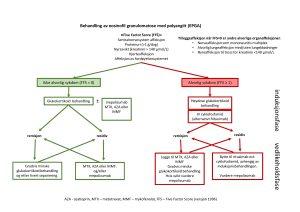
INDUKSJONSBEHANDLING
Det anbefales å innlede behandling med høydose steroid behandling ved alvorlig sykdomsbilde som oftest med IV metylprednisolon (SoluMedrol) 250-1000 mg IV x1 daglig i 3 dager. Alternativt kan man bruke per oral Prednisolon behandling 1 mg/kg (maks 60 mg/d) over 3 dager ved mindre alvorlig tilstand. Det er stor fare for residiv ved nedtrapping av per oral Prednisolon behandling, under 15 mg/d, men målet er å komme ned til 5mg/d og evt. lavere hvis mulig. Viktig å huske at lavdose Prednisolon protokollen som ble bevist i PEXIVAS studien er ikke undersøkt ved EGPA, men kun på MPA og GPA.
Tabell 2. Forslag til per oral Prednisolon nedtrapping ved induksjonsbehandling.
2a – ved alvorlig EPGA 2b – ved mindre alvorlig EPGA
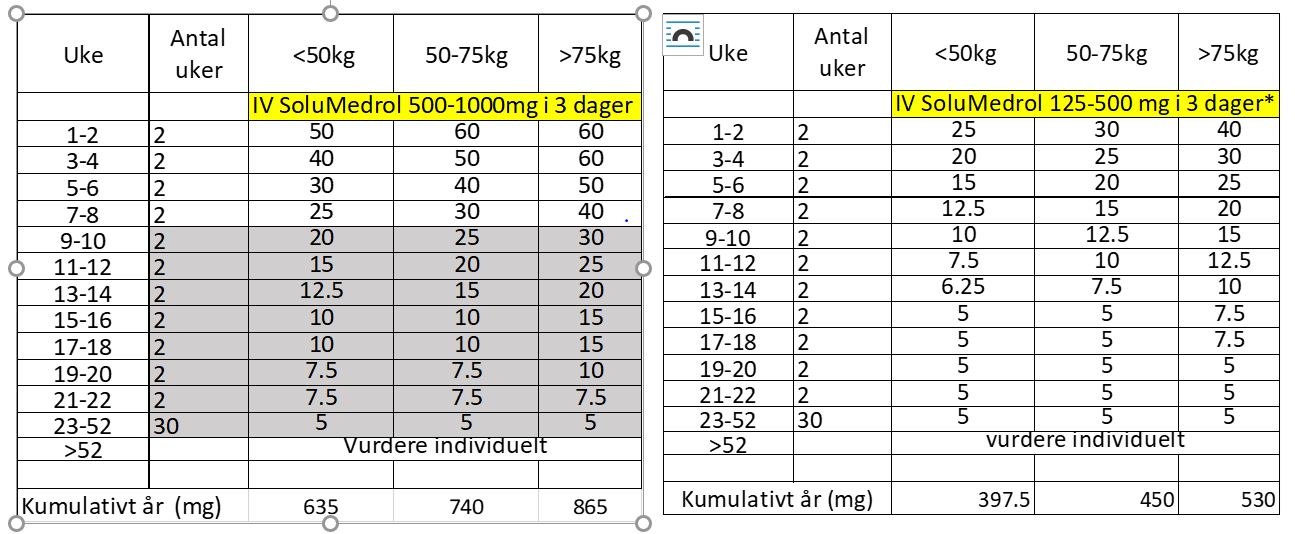
Alvorlig EGPA
Intravenøs puls behandling med cyklofosfamid, som fleste ville ha valgt, eller per oral cyklofosfamid etter EUVAS/CYCLOPS protokoll (20). Særlig ved ANCA positiv EGPA er induksjonsbehandling med IV rituksimab et alternativ. Foreløpig har man ingen randomiserte studier til å støtte dette men en rekke små ikke randomiserte studier indikerer effekt av rituksimab, der en nylig metaanalyse rapporterte 171 pasienter som fikk rituksimab enten med 375 mg/m2 per overflate ukentlig til sammen 4 ganger eller fast dose på 1000 mg med 2 ukers intervall (21). Det er gjennomført en randomisert fase III studie i Frankrike under akronymet REOVAS («Rituximab in Eosinophilic Granulomatosis With Polyangiitis») på bruk av rituksimab ved EPGA som induksjonsbehandling som inklute 108 pasienter med 1:1 randomisering, men foreløpig foreligger kun resultatene på abstrakt men er ikke blitt publiserte (NCT02807103). Det er også igangsatt en annen fase III-IV studie i Frankrike på vedlikeholdsbehandling med rituksimab på EGPA som har svært lik protokoll som tidligere studier på GPA og MPA med 500 mg rituksimab hver 6. måned. Det er 1:1 randomisering med 98 pasienter inkluderte og forventet avslutning rundt sommeren 2022. Studien går under akronymet MAINRITSEG (“Maintenance of Remission With Rituximab Versus Azathioprine for Newly-diagnosed or Relapsing Eosinophilic Granulomatosis With Polyangiitis”) (NCT03164473).
IV cyklofosfamid behandling bør vurderes hos følgende pasienter med EGPA: Hjerteaffeksjon. Ved alvorlige nevrologiske og/eller gastrointestinale manifestasjoner Og ved residiv når tidligere har vært brukt induksjonsbehandling med rituksimab.
Rituksimab behandling kan vurderes hos følgende pasienter med EGPA: ANCA positive pasienter. Ved residiv når tidligere har vært brukt induksjonsbehandling med cyklofosfamid særlig hos de få med nyreaffeksjon og glomerulonefritt.
Mindre alvorlig EPGA
Ved fredelig sykdomsbilde kan man vurdere monoterapi med steroider og vurdere tilleggsbehandling ved tilbakefall. Man kan også velge immunsupprimerende i tillegg til steroid behandling i starten til tross for at det foreligger dårlig dokumentasjon på dette. Ut fra nåværende kunnskap kan man på lik linje vil man vurderer metotreksat (MTX), azathioprin (AZA) eller mykofenolat (MMF) der resultatene av dette ser ut til å være dårlig med dessverre svært begrenset steroidsparende effekt. Hvis det er utilfredsstillende steroidsparende effekt av en av disse, der det er lite sannsynlighet at man får nevneverdig klinisk effekt at skifte mellom dem men gå rett over til anti-IL5 antistoffer.
Hos pasienter spesielt med astma med og uten sinus og nasale symptomer kan sc. mepolizumab (anti-IL-5 antistoff) vurderes også hvis pasienten får tilbakefall under redusert steroidbehandling. Det foreligger randomisert kontrollert studie som viser klar effekt av sc. mepolizumab som induksjonsbehandling på ikke alvorlig EGPA (22). Definisjonsmessig kan pasienter med EGPA og uttalt asthma også bli kategorisert som eosinofil asthma (ICD10 J82).
B: INDUKSJON, alternativ 1: CYCLOFOSFAMID (CYC) -Sendoxan®
Behandlingsprotokoll er som ved GPA og MPA. Man anbefaler IV puls behandling foran daglig per oral cyclofosfamid behandling (2mg/kg), der da foreligger lavere total kumulativ dosering av cyclofosfamid. Cyklofosfamid gis etter EUVAS/CYCLOPS protokoll med intravenøse pulser med cyclofosfamid 15 mg/kg (max 1.200 mg) med de første tre kurene med to ukers intervall (uke 0, 2, 4) og deretter med tre ukers intervall (uke 7, 10 evt. 13, 15….). Når pasienten har kommet i remisjon vil man gå over til vedlikeholdsbehandling (se Figure 1). Cyklofosfamid doseringen både må tilpasses alder samt nyresvikt (se Tabell 3).
Tabel 2 Doseendring av cyklofosfamid relatert til alder / nyrefunksjon
| CYC i.v. puls (mg/kg)
(Max 1200mg) |
||
| Alder (år) | Kreat ≤ 300(μmol/L) eller
eGFR > 30 (ml/min/1,73m2) |
Kreat > 300
eGFR: ≤ 30 |
| < 60 | 15 | 12,5 |
| 60 – 70 | 12,5 | 10,0 |
| > 70 | 10,0 | 7,5 |
B: INDUKSJON, alternativ 2: RITUXIMAB (RTX)
Rituksimab 1.000 mg i.v. gitt med 2 ukers intervall, eller
Rituksimab 375 mg/m2 overflate i.v. ukentlig i 4 uker. 30-60 min. før infusjon av rituksimab gis følgende premedikasjon:
- 1g paracetamol
- 10 mg cetrizin p.o. eller 5 mg deksklorfeniramin i.v.
- 125 mg metylprednisolon (Solu-Medrol®) i 100 ml NaCl 9 mg/ml i.v.
C: INDUKSJON alternativ 3a: METOTREKSAT (MTX).
Metotreksat p.o eller s.c. MTX kan vurderes ved begrenset EGPA, uten truende organaffeksjon. MTX s.c. er å foretrekke, for bedre og mer stabil blodverdi. 0,3 mg/kg/uke (15-25 mg), trappes opp over max. 4 uker. Det har vært tradisjon å gi samtidig po. folsyre for eksempel 1 mg daglig.
C: INDUKSJON alternativ 3b: MYKOFENOLAT (MMF) – CellCept®
Mykofenoalt mofetil kan vurderes ved begrenset EGPA uten truende organaffeksjon. Dosering: 2-3 g/d fordelt på to doseringer, med ca. 12 timers intervall. Det er tabletter på 500 mg og kapsler på 250 mg i tillegg finnes det i mixtur.
C: INDUKSJON alternativ 3c: AZATIOPRIN (AZA) – IMUREL®
Azathioprin (AZA) – Imurel® kan vurderes ved begrenset EPGA uten truende organaffeksjon
Man anbefaler å ta. tiopurin methyltransferase (TPMT) genotype forut behandling. De fleste (rundt 90%) har TPMT*1/*1 (wild-type) genotype med normal TPMT enzym aktivitet og kan få vanlig dosering, 2,0-2,5 mg/kg per dag en gang på dagen. Pasienter som er heterozygote med TPMT*1 og noen av de over 20 TPMT polymorfismene, som oftest er *2, *3A, *3B, *3C, og *4, har redusert TPMT enzym aktivitet og må få redusert, oftest halvert dosering og følges grundig hvis oppstart. De som ikke har TPMT*1genotypen og enten homozyt og/eller heterozygot for en av de allelene bør ikke få azathioprin pga. økt risiko for alvorlig myelosuppresjon. Dette utgjør få pasienter (>1%). Vises til Avdeling for farmakologi. Oslo universitetssykehus. https://anx.no/tpmt/
Verdi av 6-tioguaninnukleotide (6-TGN) og metyl-merkaptopurin (me-MP) måles i heparinisert fullblod, tilsier om effekt av AZA behandlingen. Terapeutisk område er: 6-TGN 3,5−5,0 µmol/L og me-MP <50 µmol/L når brukes i transplantasjonsmedisin. Dette kan anvendes i kontroll med blodprøve. Se https://anx.no/6tgn/
Imurel finnes i 50 og 25 mg tabletter.
C: INDUKSJON alternativ 3d: Anti-Interleukin 5 antistoffer
MEPOLIZUMAB – NUCALA®
Mepolizumab, anti–interleukin-5 monoklonal antistoff behandling er registret i Norge for behandling av; EGPA og HES samt for terapiresistent eosinofil astma (ICD10 J82) og kronisk rhinosinusitt med nasal polypose (CRWwNP).
Mepolizumab er her i Norge til i 100 mg ferdigfylt penn eller sprøyte og som 40 mg i ferdigfyllt sprøyte.
Det er publisert en fase III, randomisert dobbeltblindet kontroll studie (MIRRA trial) som viste at behandling med mepolizumab 300 mg sc. hver 4. uke, gitt i tillegg til steroid behandling hos pasienter med ikke alvorlig EGPA, var bedre enn placebo. I tillegg var dokumentert steroidsparende effekt (22). Mepolizumab har bra bivirkningsprofil, men foreløpig høy pris. Dokumentasjonen på EGPA er basert på induksjonsbehandling, men mepolizumab kan også brukes i vedlikeholdsfasen, men der har man ikke randomiserte kontrollerte studer. Mulighet er å bruke dette «off label» hos pasienter som er som ikke klarer å trappe ned steroidbehandling, og dessverre er det svært dårlig effekt av annen immunsuppresjonsbehandling (MTX, AZA, MMF, RTX & CYC ). Man kan vurdere å bruke samme behandlingsstrategi og ved eosinofil astma med oppstart av 100mg sc. hver 4. uke og evt. vurder økt dosering til 200 evt. 300mg hvis manglende respons med målet å komme ned i per oral prednsiolon dosering under 7.5mg/d og hvis mulig under 5 mg/d og evt. seponering. Problemstillingen ved denne behandlingen er pris og her må man i flestalle tilfeller legge seg på langvarig behandling.
Nylig er publisert studie (MANDARA trial) som sammenligner to typer av anti-IL-5 antistoffer, mepolizumab (300mg sc hver 4 uke) og benralizumab (30mg sc. hver 4. uke), i en randomisert dobbeltblindet kontroll studie i 52 uker, med total 140 pasienter med ralapserende eller refraktær EGPA med 70 pasienter i hver gruppe. Resultatet viste at benralizumab var ikke dårligere (non-inferiority) men ikke statistisk bedre (non-superiority) til tross at flere i benralizumab behandlingsgruppen, 41% vs 26% klarte å fase helt ut Prednisolon og at benralizumab medførte til gjennomsnittlig lavere eosinofiltall sammenlignet med de som fikk mepolizumab (29).
BENRALIZUMAB – FANSENRA®
Benralizumab er anti–interleukin-5 monoklonal antistoff. Foreløpig er benralizumab kun registrert i Norge på alvorlig asthma. Det finnes i ferdigfyllt pen eller sprøyte på 30 mg og gis hver 4. uke.
Problemstillingen ved denne behandlingen er pris og her må man i flest alle tilfeller legge seg på langvarig behandling og ut fra dagens prissetning er 30mg Fansenra® fra AstraZenica litt dyrere en 3 x100mg Nucala® fra GlaxoSmithKline og ligger på rundt 410.000Nkr året mens 100mg Nucala koster rundt 144.000 Nkr året. Man har som oftest startet opp med Nucala 100mg sc. uken og økt doseringen hvis nødvendig og de som ikke klarer å redusere Prednisolon doseringen under 7.5mg/d-5mg/d. Når prisen på disse medikamenter faller vil man forvente tidligere og mer utstrakt bruk.
Prognose ved EGPA
Prognosen for pasienter med EGPA har forbedret seg betydelig siden bruk av systemiske glukokortikoid behandling ble utbredt og senere innføring av steroidsparende behandling i hovedsakelig cyklofosfamid ved alvorlig sykdom. Det har vært funnet sammenheng med «five-factor score» og overlevelse og der er ikke overaskende alder > 65 år assosiert til økt mortalitet i tillegg til hjerte- og gastrointestinal affeksjon. I en tysk studie på EGPA med 104 inkluderte pasienter var 10-år overlevelse på 89% som ikke var særlig lavere enn i generell befolkning, der standardisert mortalitet rate (SMR) lå på 1,29 (23). En nylig japansk studie som inkluderte 188 pasienter viste en 5-år overlevelse på 89,6% år og en 5 år relapse-fri overlevelse på 64% (24).
Vedvarende og langvarig steroid behandling som dessverre stor del av pasientene er avhengig av relateres til morbiditet, permanent skade i form av økt risiko for hjertekarsykdom, økt risiko for diabetes mellitus og overvekt samt for osteoporose og katarakt og leder til økt mortalitet over tid. Forhåpentlig kan økt bruk av effektive steroid sparende behandlingsalternativer med bla. hemming av interleukin 5, der man nå har flere behandlingsalternativer. Per dags dato har man i tillegg til mepolizumab som foreløpig er best utredet for EGPA, har man benralizumab (25-27) som viser i hvert fall ikke dårligere effekt en mepolizumab men problemstillingen her er relativ høy pris av anti-IL5 antistoff behandlingen.
Anbefalte oversiktsartikler om EGPA
Referanser
- Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
- Lyons PA, Peters JE, Alberici F, Liley J, Coulson RMR, Astle W, et al. Genome-wide association study of eosinophilic granulomatosis with polyangiitis reveals genomic loci stratified by ANCA status. Nat Commun. 2019;10(1):5120.
- Churg J, Strauss L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol. 1951;27(2):277-301.
- Kotzin JJ, Spencer SP, McCright SJ, Kumar DBU, Collet MA, Mowel WK, et al. The long non-coding RNA Morrbid regulates Bim and short-lived myeloid cell lifespan. Nature. 2016;537(7619):239-43.
- Nguyen Y, Guillevin L. Eosinophilic Granulomatosis with Polyangiitis (EGPA, Churg–Strauss). In: Sinico RA, Guillevin L, editors. Anti-Neutrophil Cytoplasmic Antibody (ANCA) Associated Vasculitis. Rare Diseases of the Immune System. Cham: Springer International Publishing; 2020. p. 77-95.
- Watts R, Lane S, Hanslik T, Hauser T, Hellmich B, Koldingsnes W, et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis. 2007;66(2):222-7.
- Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore). 1984;63(2):65-81.
- Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990;33(8):1094-100.
- Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, Emery P, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM. 1994;87(11):671-8.
- Mukhtyar C, Lee R, Brown D, Carruthers D, Dasgupta B, Dubey S, et al. Modification and validation of the Birmingham Vasculitis Activity Score (version 3). Ann Rheum Dis. 2009;68(12):1827-32.
- Exley A, Bacon P, Luqmani R, Kitas G, Gordon C, Savage C, et al. Development and initial validation of the Vasculitis Damage Index for the standardized clinical assessment of damage in the systemic vasculitides. Arthritis Rheum. 1997;40:371 – 80.
- Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P. The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore). 2011;90(1):19-27.
- Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore). 1996;75(1):17-28.
- Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum. 2013;65(1):270-81.
- Hellmich B, Sanchez-Alamo B, Schirmer JH, Berti A, Blockmans D, Cid MC, et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Ann Rheum Dis. 2024;83(1):30-47.
- Maric I, Sun X. Advances in diagnosis of mastocytosis and hypereosinophilic syndrome(☆). Seminars in hematology. 2019;56(1):22-9.
- Shomali W, Gotlib J. World Health Organization-defined eosinophilic disorders: 2019 update on diagnosis, risk stratification, and management. Am J Hematol. 2019;94(10):1149-67.
- Iurlo A, Cattaneo D, Gianelli U. Hypereosinophilic syndromes in the precision medicine era: clinical, molecular aspects and therapeutic approaches (targeted therapies). Expert review of hematology. 2019;12(12):1077-88.
- Chung SA, Langford CA, Maz M, Abril A, Gorelik M, Guyatt G, et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2021;73(8):1366-83
- Mukhtyar C, Guillevin L, Cid M, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of primary small vessel vasculitis. Ann Rheum Dis. 2009;68:310 – 7.
- Akiyama M, Kaneko Y, Takeuchi T. Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis: A systematic literature review. Autoimmun Rev. 2020:102737.
- Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford CA, et al. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. N Engl J Med. 2017;376(20):1921-32.
- Moosig F, Bremer JP, Hellmich B, Holle JU, Holl-Ulrich K, Laudien M, et al. A vasculitis centre based management strategy leads to improved outcome in eosinophilic granulomatosis and polyangiitis (Churg-Strauss, EGPA): monocentric experiences in 150 patients. Ann Rheum Dis. 2013;72(6):1011-7.
- Saku A, Furuta S, Hiraguri M, Ikeda K, Kobayashi Y, Kagami SI, et al. Longterm Outcomes of 188 Japanese Patients with Eosinophilic Granulomatosis with Polyangiitis. J Rheumatol. 2018;45(8):1159-66.
- Coppola A, Flores KR, De Filippis F. Rapid onset of effect of benralizumab on respiratory symptoms in a patient with eosinophilic granulomatosis with polyangiitis. Respir Med Case Rep. 2020;30:101050.
- Miyata Y, Inoue H, Homma T, Tanaka A, Sagara H. Efficacy of Benralizumab and Clinical Course of Igg4 on Eosinophilic Granulomatosis with Polyangiitis. Journal of investigational allergology & clinical immunology. 2020:0.
- Nanzer AM, Dhariwal J, Kavanagh J, Hearn A, Fernandes M, Thomson L, et al. Steroid-sparing effects of benralizumab in patients with eosinophilic granulomatosis with polyangiitis. ERJ open research. 2020;6(4).
- Grayson PC, Ponte C, Suppiah R, Robson JC, Craven A, Judge A, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic Granulomatosis with Polyangiitis. Ann Rheum Dis. 2022;81(3):309-14.
- Wechsler, M. E., et al. (2024). “Benralizumab versus Mepolizumab for Eosinophilic Granulomatosis with Polyangiitis.” N Engl J Med 390(10): 911-921.
